


Introduction
Glaciers are excellent archives of atmospheric constituents. However, in order to interpret these records the atmospheric transfer function and the effect of post-depositional processes need to be known. Being reversibly deposited in the snow, the transfer function for nitrate (NO3 −) involves multiple post-depositional processes including its re-emission to the atmosphere as nitric acid (HNO3) and nitrous oxides (NOx). The loss of NO3 − from the snowpack has confounded the interpretation of a number of ice-core records. For example, Antarctic ice cores from Dome C, Vostok and the Dominion Range display NO3 − records that decrease in concentration with depth. This type profile was originally thought to reflect some change in atmospheric chemistry, but was later shown to be the result of NO3 − reemission from the snow (Reference Mayewski and LegrandMayewski and Legrand, 1990; Reference De Angelis, Legrand and DelmasDe Angelis and Legrand, 1995; Reference Wolff, Wagenbach, Pasteur, Mulvaney, Legrand and HallWolff and others, 1998). Nitrate is deposited as HNO3, which can be released from the snow surface as HNO3 gas or by conversion to NOx by photolysis (Wolff and others, 2002). Gas-phase HNO3 within the snow undergoes reversible exchange with the atmosphere and should eventually equilibrate with atmospheric levels. This phenomenon seems to be typical for low-accumulation snowfields (Reference De Angelis, Legrand and DelmasDe Angelis and Legrand, 1995). In high-accumulation snowfields there may be insufficient time for the surface snow to reach equilibrium before it is buried by new snow (Reference Bales, Choi, Wolff and BalesBales and Choi, 1996). However, as observed in Greenland, NO3 − may escape from the surface snow layers within a few days of deposition. This suggests post-depositional processes may also be important at sites with higher accumulation rates, given the right conditions (Reference Ginot, Kull, Schwikowski, Schotterer, Pouyaud and GäggelerGinot and others, 2001). The rate of conversion of HNO3 to NOx by photolysis is primarily driven by the actinic flux and the snow accumulation rate (Wolff and others, 2002). The site studied in this investigation was in a shadowed area with a relatively high snow accumulation rate. The loss of nitrate by photolysis is therefore believed to be negligible.
To investigate the effects of depositional and post-depositional processes on NO3 − at an alpine glacier site, aerosol, surface snow and snow–firn pack NO3 − was analyzed over the course of 2years. The investigation was undertaken as part of the Program for Glacier Processes Investigations (PGPI), which was initiated in July 2002 by the Tien Shan Glaciological Station (TGS). To our knowledge, this investigation was the first of its kind in the eastern Tien Shan.
Sampling Site
As part of the PGPI, an observation site (PGPI site) was established on the east branch of Ürümqi glacier No. 1 (UG1; 43.05˚ N, 86.49˚E; 4130 ma.s.l.), in the eastern Tien Shan of northwestern China (Fig. 1). This site was within the percolation zone and situated approximately 3km southwest of the Daxigou meteorological station (3539 ma.s.l.), where aerosol samples were collected. The glacier is located at the headwaters of the Ürümqi river and is surrounded by barren rock and sparsely vegetated frozen ground. The mean annual precipitation at snowline elevation (approximately 4055 ma.s.l.) is 646 mma−1, with 90% of the precipitation occurring in summer (May–September). The depth of the snow–firn pack at the site typically ranged from 1.5 m in late summer to 3 m in late spring and incorporated 3–4 years of net snow accumulation. The floor of the snow–firn pack was composed of superimposed ice, a clear and impermeable opaque ice with spherical bubbles approximately 1–5mm in diameter. The atmospheric aerosol, glacier surface snow and snow–firn pit samples were collected concurrently on a weekly basis.

Fig. 1. Geographic environment around the eastern Tien Shan (modified from Reference Lee, Qin, Jiang, Duan and ZhouLee and others, 2003) showing the proximity of the study site to deserts and gobis and the city of Ürümqi. The stippled areas designate deserts and gobis drawn on the same scale as the surrounding areas.
Atmospheric transport over the glacier typically results from the prevailing westerly circulation pattern. However, from March to September, distinctive diurnal valley winds (Reference Zhang, Kang and LiuZhang and others, 1994) may transport air up the Ürümqi river basin to the glacier. The main pollution sources in the river basin include road traffic, the city of Ürümqi and the town of Houxia, which is located near two coal power plants and a cement factory. The city of Ürümqi, the capital of the Xinjiang Uygur Autonomous Region and a large industrial city, lies 100 km to the northeast of the glacier, and Houxia (a small town) is located 50km northeast.
Data and Methods
Fifty-five total suspended particle (TSP) aerosol samples were collected using 47 mm diameter ZefluorTM (2μm pore size) Teflon filters (Gelman Sciences), from 27 December 2002 to 21 February 2004 on a weekly basis. Filtered air volumes were measured by an in-line flow-meter and converted into standard cubic meters (m3) using ambient temperature and pressure data. The particle collection efficiency (for particles as small as 0.035 μm) was estimated to be >97% based on the mean flow rate of 1.27 m3 h−1 over the filter (Reference Liu, Pui, Rubow, Marple and LiuLiu and others, 1983). Strict trace-chemistry procedures were followed during sampling and transportation to prevent contamination both in the field and the laboratory, including using disposable polyethylene gloves, oronasal masks and pre-cleaned polyethylene sample containers. The filter cartridges were loaded and packed into clean plastic bags in a class 100 clean laboratory at the TGS and transported to the field in airtight containers. After sampling, the filters were removed from the cartridges and placed in pre-cleaned, airtight glass bottles. Blank filters were handled in the same manner as the samples. To analyze the major-ion concentrations, the sample and blank filters were wetted by 0.2 mL ultrapure alcohol. The soluble components were then extracted with 25 mL aliquots of deionized Milli-Q water. The detection limits for NO3 − in aerosol were 0.18 nEq m−3. Detailed sampling methods and analytical techniques of aerosol are described by Reference Shrestha, Wake and DibbShrestha and others (1997, Reference Shrestha2000) and Reference Zhao and LiZhao and Li (2004).
A total of 107 surface snow samples (usually 1–5cm of uppermost snow) were collected at the PGPI site at 7day intervals from 14 September 2002 to 28 September 2004. The surface snow consisted mainly of fresh snow during summer (the wet season) and of relatively old snow during winter because of sporadic precipitation (dry season). During surface sampling an effort was made to collect the fresh snow and well-preserved surface snow (i.e. snow that was not affected by post-depositional processes such as sublimation or melting) at the sampling site each week to investigate the chemical seasonality in precipitation and dry deposition. Therefore, during the winter season when there was insufficient snowfall, we usually sampled the topmost 3 cm. However, if an accumulation event occurred prior to the sampling, we collected the top 1 cm of the fresh snow. During the summer season, there was usually sufficient fresh snow available, and samples from accumulation that was no more than 2days old were usually collected from the top 3–5cm.
A snow–firn pit was concurrently sampled from top to bottom with 10cm resolution to yield a total of 1011 samples from September 2003 to September 2004. The snow-pit wall was scaled back by at least 50cm before the collection of samples, and the pit was refilled after each sampling campaign.
Snow samples were kept frozen in the field, during transportation and in the laboratory before the analysis. The analysis of duplicate samples as well as field and laboratory blanks indicates that sample contamination during sample collection, transport and subsequent analytical procedures was negligible. Both aerosol and snow samples were analyzed in the TGS laboratory, using a Dionex Ion Chromatograph model DX-320. Detailed methods are described by Reference Buck, Mayewski, Spencer, Whitlow, Twickler and BarrettBuck and others (1992), Reference Wake and MayewskiWake and Mayewski (1993) and Reference Zhao and LiZhao and Li (2004).
Results and Discussion
Variations in aerosol and surface snow
Aerosol and surface snow NO3 − concentrations are shown in Figure 2. The mean aerosol NO3 − concentration during the study was found to be 6nEqm– 3 with minimum concentrations below the detection limit (0.18 nEq m−3) and a maximum concentration of 31.3 nEq m−3. Seventy-four per cent of the values were found to be <15 nEq m−3. The aerosol NO3 − concentrations were found to exhibit some seasonal variability, with mean values for spring, summer, autumn and winter of 3.9, 5.0, 13.0 and 4.7 nEq m−3, respectively. The high aerosol NO3 − concentrations during autumn most likely resulted from polluted air masses being transported up the Ürümqi river basin by valley winds (Reference Lee, Qin, Jiang, Duan and ZhouLee and others, 2003).

Fig. 2. NO3 − concentrations and their variation trends in aerosol and surface snow samples collected on UG1. The smoothed curve is generated from negative exponential smoothing with sampling proportion 0.1 and polynomial degree 1. Dates are mm/dd/yy.
Surface snow NO3 − concentrations (Fig. 2) varied from extremely high levels (>2000ngg−1) from mid-September to the end of October 2002, to concentrations below 500 ng g−1, with little variability until the beginning of April 2003. Sharp peaks in surface snow nitrate occurred on 5 April 2003, and from mid-May to the end of August 2003 and then once again remained relatively constant below 500 ng g−1 until the end of January 2004. The period February–October 2004 was characterized by successive high peaks with a short, stable period during April and May. Clearly the most significant feature of the surface snow concentration NO3 − record occurred from mid-September to the end of October 2002, with initial NO3 − concentrations >4000ngg−1. Air-mass back trajectories (48 hours) calculated using the US National Oceanic and Atmospheric Administration (NOAA) Hyspit model and European reanalysis (ERA-40) data suggest that these extremely high concentrations occurred during atmospheric transport to the site from large late-season fires, which occurred in the steppes of Kazakhstan to the east.
During the observation period, the seasonal variability in surface snow NO3 − was relatively ambiguous. The average concentrations of NO3 − in spring and summer were very similar: 390.9 and 392.4 ngg−1, respectively. In autumn the value was much higher than in the other seasons (mean of 657 ng g−1); however, the mean concentration for this season is somewhat biased by the high concentrations found during 2002. During winter, NO3 − appears to be relatively stable, with lowest average value of 348 ng g−1. The mean concentration for all samples was found to be 446.4 ng g−1 (7.2μEq L−1).
Relationship between NO3 − in surface snow and aerosol
The atmospheric flux of NO3 − to the Earth surface occurs by both dry and wet deposition processes. Both types of deposition strongly depend on meteorological parameters (e.g. precipitation, wind). Surface snow samples collected in summer (wet season) consisted mainly of fresh snow, while winter surface snow samples were relatively old because of sporadic precipitation (dry season). The impact of dry deposition and post-depositional processes, such as wind erosion, sublimation and condensation, should be greater for the winter surface snow compared to the summer due to the low precipitation rate. A significant linear relationship (R 2 = 0.89, N = 10, p< 0.01) between NO3 − concentrations in the surface snow and aerosol was found during the winter (December 2003 to mid-February 2004; Fig. 3). No correlation between aerosol and surface snow NO3 − was found for the spring and summer. Similar relationships between aerosol and surface snow NO3 − have been observed at other alpine sites (e.g. Reference BaltenspergerBaltensperger and others, 1993). The correlation between aerosol and surface snow NO3 − during the winter is most likely due to dry deposition processes and a longer equilibration time with the atmosphere. In contrast, wet deposition processes are thought to dominate in the summer, due to a much greater precipitation rate. The NO3 − concentration in the fresh surface snow is determined by two sources: NO3 − within precipitation and NO3 − scavenged from air by precipitation. Low concentrations of NO3 − in aerosol during the wet season suggest a scavenging effect, i.e. an efficient removal of aerosol particles during persistent precipitation, possibly resulting in significant dilution of NO3 − and HNO3 in air and enrichment of those in snow. This might have happened on the sampling days 8 May, 26 June and 12 July 2003. On these dates the NO3 − concentrations in aerosol were too low to be detected, possibly because the samples were collected after a long period of continuous precipitation.

Fig. 3. Comparison of NO3 − in surface snow with aerosol, and linear regression calculated for period December 2003 to mid-February 2004. Dates are mm/dd/yy.
Despite the low correlation between the concurrently collected aerosol and surface snow samples in the spring, summer and autumn, the NO3 − concentration in the aerosol evidently shows a trend similar to that in surface snow (Fig. 2), suggesting that the low-frequency variation trend of atmospheric NO3 − may have been preserved in the surface snow. The relationship between NO3 − in the aerosol and snow during the higher precipitation months is evidently very complex and may be non-linear due to a disequilibrium between the air and snow or vertical NO3 − gradients in the air above the area.
Evolution processes in snow–firn
During the investigation it was observed that the surface snow usually had higher NO3 − concentrations than older snow collected below the surface from a snow pit. This is thought to be largely the result of the NO3 − eluting downwards in the warmer months from percolation within the snowpack. In order to investigate the relative mobility of NO3 − it was compared with Mg2+, which is primarily derived from mineral aerosol particles in the snow. Figure 4 shows the evolution process of NO3 − in the snow–firn pack at the site from October 2003 to September 2004. Two prominent NO3 − peaks (P1 and P2) in the snow pit at the beginning of October 2003 were tracked through the snowpack over the course of the investigation. The lower peak (P1) was formed before mid-September 2003, and the upper peak (P2) was found to be shaped by intense precipitation at the end of September 2003. From mid-November 2003 to late April 2004, the positions of P1 and P2 remained relatively stable, with a slight sinking towards the superimposed ice surface, which could be caused by snow compaction. Several small peaks of concentration in the upper parts of the snow pits appear as the result of limited precipitation, low temperatures and weak evaporation, which enabled post-depositional processes such as sublimation and dry deposition to become more dominant.

Fig. 4. The evolution process of vertical profiles of NO3 − and Mg2+ in snow pits from 4 September 2003 to 8 September 2004. P1–P3 indicate NO3 − concentration peaks, and P10 and P20 indicate Mg2+ concentration peaks. Height (cm w.e.) is above the superimposed ice surface. Dates are mm/dd/yy.
From April to the end of May, as temperature increased, meltwater appeared in the snow pit, which attenuated the peaks somewhat as they moved downward. At the same time, the increasing precipitation resulted in successive concentration peaks at about 10–30 cm below the surface. In the summer (June–August), as meltwater percolated through the snow–firn pit, it leached ions, including the NO3 − and Mg2+. During this period, the upper part of the pit experienced the most post-depositional alteration. With increasing temperature, larger volumes of meltwater began to percolate deeper into the pit along with the NO3 − and Mg2+, which is indicated by displacement and reduction in concentration of the peaks. When meltwater encountered the superimposed ice at the bottom of the coarse firn layer, it refroze, resulting in the rise of the superimposed ice surface. The extensive ablation caused the compaction of the concentration peaks. At the end of August, P1 merged into the superimposed ice at the bottom of the pit and was henceforth preserved in the glacier as a new annual layer. This demonstrates that in spite of considerable loss of NO3 − and Mg2+ through post-depositional processes, the general information is still preserved in the snow pit. Around mid-August, a new peak (P3) emerged in the upper layer of the pit, and quickly sank downwards.
In comparison with Mg2+, the NO3 − appeared to be preferentially leached downward in the snow pit. This is clearly illustrated during the period June–August 2004 as elution intensified. On 25 June, both P1 and P2 were located above P10 and P20 (the two peaks were traced in Mg2+ concentration profiles) in the pit, but in the profiles of 25 August both P1 and P2 were found in positions below P1’ and P2’ , which indicated that NO3 − was more efficiently leached downward. The relatively slow elution of Mg2+ is probably due to some of the Mg2+ being bound to dust layers in the snow–firn pit.
Spatial comparisons
Mean NO3 − concentrations in snow from different glaciers are shown in Table 1. Antarctic data are generally lower than 1.6 μEq L−1, with slightly higher concentrations reported for Greenland. The lowest reported snow NO3 − level was found at East Rongbuk Glacier during the monsoon season (northern slope of Qomolangma). The highest concentrations in East Rongbuk Glacier occur in spring (pre-monsoon season), resulting from dust aerosol input during dust storms (observed mainly in April and May in western China), and the lowest concentrations occur in late summer (monsoon season). The lower summer concentrations reflect not only decreased dust deposition, but also the effect of elution (Reference Kang, Mayewski, Qin, Sneed, Ren and ZhangKang and others, 2004). Compared to these sites, the mean concentration of NO3 − in UG1 is much higher. This may be attributed to the transport of terrestrial dust, biomass burning and anthropogenic emissions in the region. Further research is required to investigate the sources of NO3 − to the eastern Tien Shan and also whether aquatic ecosystems are being impacted.
Table 1. Comparison of NO3 − in surface snow with other remote glacier areas

With respect to UG1, the concentrations of NO3 − in Inilchek glacier (42˚12.5’ N, 80˚12.2’ E), central Tien Shan, provide a comparable dataset. Table 2 presents seasonal NO3 − values of Inilchek glacier from a firn–ice core recovered in 1998 from the accumulation zone at 5100 m a.s.l. (Reference Aizen, Aizen, Melack, Kreutz and CecilAizen and others 2004).
Table 2. Seasonal average values of NO3 − in UG1 and Inilchek glacier
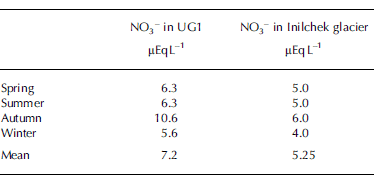
While the seasonal variations of NO3 − in UG1 and Inilchek glacier are similar, the values of NO3 − in UG1 are generally higher. On Inilchek glacier, the changes in NO3 − in spring and summer were associated with increasing frequency of northwestern advection, which originated over the southern Urals and passed through western Kazakhstan. Both western Kazakhstan and the southern Urals are industrial areas and potential sources of pollution (Reference Aizen, Aizen, Melack, Kreutz and CecilAizen and others, 2004). For both areas, anthropogenic pollution, including emissions from fossil fuel combustion and biomass burning, livestock manure, and commercial and natural fertilizers, may be the principal source of NO3 −. Mineral dust acts as a carrier for these pollutants (Reference Li, Yao and XieLi and others, 1995, Reference Li, Lu, Liu and Fu1999) and may be partially responsible for high levels of NO3 − in these two areas, since both areas are impacted by regional and local Asian dust.
Summary
A 2 year study of aerosol, surface snow and snow-pit NO3 − was conducted to determine the depositional and post-depositional processes that occur on UG1. The concentrations of NO3 − in both surface snow and aerosol display ambiguous seasonal changes. Low values are commonly observed during winter, and relatively high concentrations during autumn. Extremely high NO3 − (>4000ngg−1) concentrations were found in surface snow during September and October 2002. These concentrations are high enough to have a significant impact on the aquatic ecosystems of the eastern Tien Shan. Atmospheric back trajectories suggest that this event may have been the result of large late-season fires in Kazakhstan. Other possible sources of the NO3 − include local-to-regional anthropogenic emissions, which may be enhanced by transport in dust storms. Further research is required to assess whether such events are impacting the aquatic ecosystems of the region. A significant correlation between aerosol and surface snow NO3 − concentrations was found for the dry season from December 2003 to mid-February 2004. The dominance of dry deposition during this season is believed to be the primary cause of the high correlation. While there was no significant correlation during the other seasons, a similar trend in the aerosol and surface snow NO3– was found, suggesting that long-term variations in atmospheric NO3 − may be preserved in the surface snow. From the analysis of snowpack NO3 − profiles it was determined that the NO3 − stratigraphy at the site is formed by different processes during winter and summer. During winter the NO3 − stratigraphy is due to the dry deposition of ambient aerosol followed by snow compaction and sublimation. During summer, NO3 − is deposited primarily by wet deposition and then eluted downward through the snowpack by meltwater. The percolation of meltwater was found to attenuate the NO3 − stratigraphy, but still preserved some of its temporal variability in superimposed ice at the bottom of the snowpack. However, co-registration of NO3 − with other soluble chemical species within the superimposed ice is probably lost due to different elution characteristics. This has many implications for the interpretation of chemical records in similar alpine glaciers.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (40121101; 40371028; 40571033; J0130084) and the Chinese Academy of Sciences (CAS) and the Cold and Arid Regions Environmental and Engineering Research Institute (KZCX3-SW-341; CACX2003101). We also thank V. Aizen, D. Joswiak and R. March for useful comments and helpful suggestions. Support for this research has been provided under the Program for Glacier Processes Investigation conducted by the Tien Shan Glaciological Station, CAS.






