


Introduction
Antarctica’s role in the global climate system is both pivotal and complex. West Antarctica in particular is the most dynamic area of the continent both atmospherically and glaciologically (Reference Cullather, Bromwich and Van WoertCullather and others, 1996; Reference BindschadlerBindschadler, 1998). It is impacted by several major atmospheric weather systems, the Amundsen Sea low and, to a lesser degree, the Weddell Sea low and the Davis Sea low. These large atmospheric low-pressure systems are the primary transport mechanisms for moisture and aerosols to the West Antarctic ice sheet.
Instrumental climate records are relatively sparse over the Southern Hemisphere and extend back <100 years. Over Antarctica such records extend back only a few decades. Glaciochemical proxy data can extend the paleoclimate record back hundreds to thousands of years and also provide a unique resource for examining changes in the sources, pathways and distribution of chemical species in the atmosphere through time (Reference MayewskiMayewski and others, 1993). This paper focuses on the sulfate (SO4 2–) time series available from a series of 16 ice cores collected over West Antarctica.
Sulfate aerosols play a significant role in the heat budget of the global atmosphere, mainly through the scattering of incoming solar radiation and through indirect effects involving clouds (Reference Charlson, Langner and RohdeCharlson and others, 1990). In addition to anthropogenic emissions, sulfur is released into the atmosphere from a variety of natural sources including sea salt, continental dust, volcanic eruptions, the terrestrial biosphere and the marine biosphere.
Major volcanic events such as the eruption of Pinatubo, Philippines, in 1991 can inject large volumes of sulfur gases (H2S, SO2), ash, dust and crustal material directly into the stratosphere. Most large volcanic eruptions (such as Krakatau, Indonesia, in 1883 and Agung, Indonesia, in 1963) significantly increase stratospheric SO4 2– concentrations for at least 1–2 years after the event. Occasionally, an exceptionally large eruption, such as Tambora, Indonesia, will increase atmospheric SO4 2– concentrations for 3–4 years (Reference Self, Rampino and BarberaSelf and others, 1981). Large volcanic eruptions significantly affect stratospheric chemistry, inducing a higher catalytic destruction rate of ozone, resulting in enhanced levels of ultraviolet range B (UV-B) radiation at the Earth’s surface (Reference Berresheim, Wine, Davis and Singh, H.B.Berresheim and others, 1995).
Antarctica is an ideal place to study natural atmospheric SO4 2– variability due to its isolated location, the fact that Antarctic precipitation is an excellent repository for the deposition of soluble and insoluble chemical species, and its remoteness from major anthropogenic SO4 2– sources that can confound the investigation of natural variability over more populated regions (Reference ShawShaw, 1982; Reference LegrandLegrand and Mayewski, 1997). The primary types of SO4 2– in Antarctic aerosols are sea-salt (ss) SO4 2– and excess (xs) SO4 2–, the latter composed predominantly of marine biogenic emissions, volcanic emissions (Reference DelmasDelmas, 1982; Reference LegrandLegrand, 1997) and an as yet either non-existent or undetermined contribution from anthropogenic activity. Marine biogenic emissions dominate the overall sulfur budget in the Southern Hemisphere (Reference Bates, Lamb, Guenther, Dignon and StoiberBates and others, 1992; Reference Legrand and MayewskiLegrand and Mayewski, 1997).
In this study, 16 sub-annually resolved, continuously sampled, multivariate ice-core records (15 from West Antarctica and 1 from South Pole; Fig. 1) are used to investigate ice-core SO4 2– concentrations over West Antarctica. The 16 ice cores capture the sub-annual variability in Antarctic SO4 2– loading over the period 1487–2002, with a ∼200 year overlap (1799–1992) for six of the cores, a 101 year overlap (1891–1992) for 11 of the cores and a 50 year overlap (1952–92) for all cores except 01-6 (Table 1).
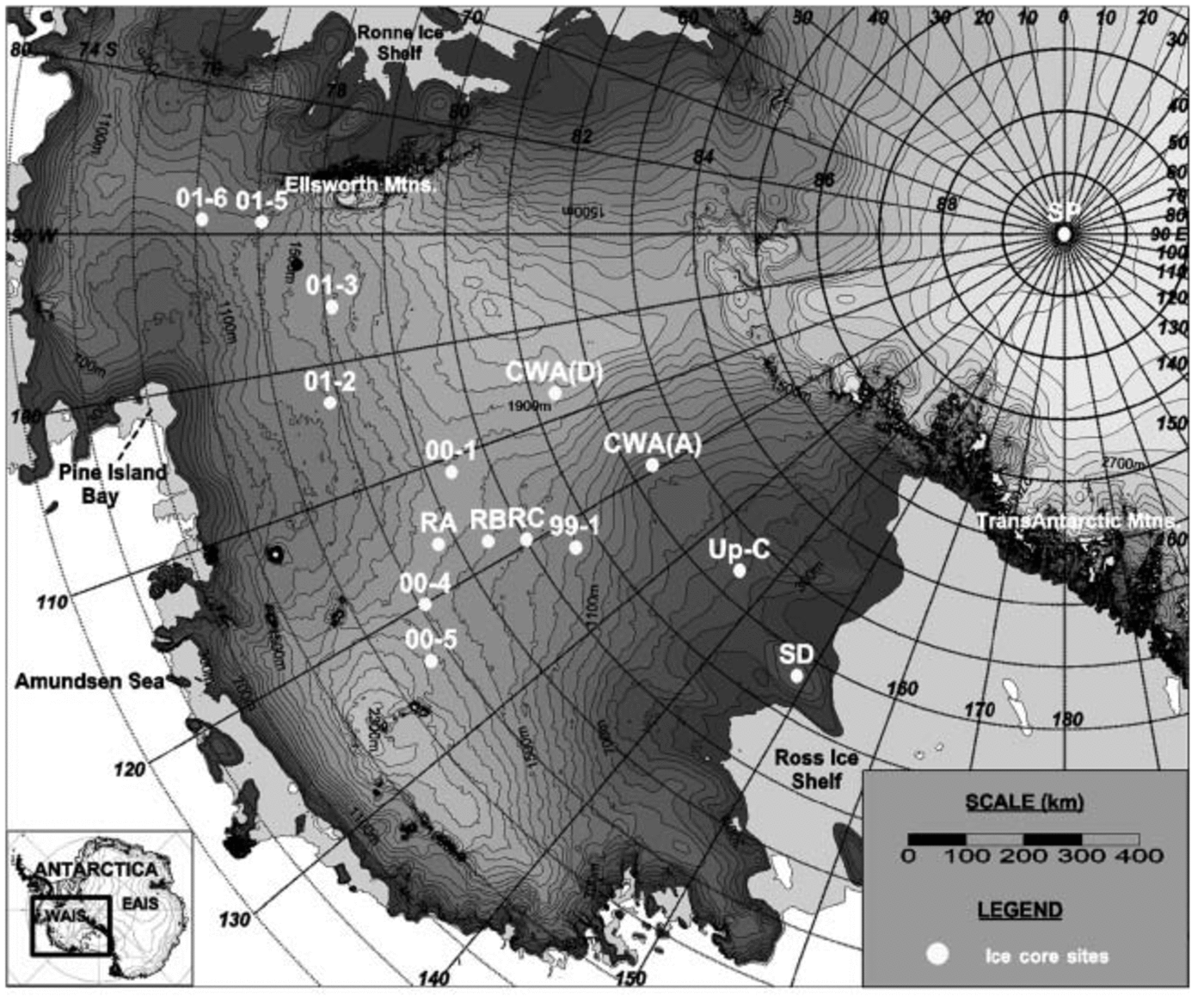
Fig. 1. Location map of sites for all ice cores used in this study. RA, RB and RC represent core sites RIDS-A, RIDS-B and RIDS-C, respectively. Map created using the RADARSAT-1 Antarctic Mapping Project (RAMP) digital elevation model (Reference Liu, Jezek, Li and ZhaoLiu and others, 2001).
Each ice-core SO4 2– time series is separated into its primary constituents ssSO4 2– and xsSO4 2–, from which the spatial and temporal variation of these fractions is investigated for the period of the last 200 years. The methods used for separating ss from xs are discussed later. Volcanic peaks are extracted from the xsSO4 2– time series by calculating the residuals from a robust-spline smoothing of the raw xsSO4 2– data following the technique used by Reference ZielinskiZielinski and others (1994). Several well-documented volcanic events are detected in each xsSO4 2– series and these are used to reinforce the identification of annual-layer dating by providing absolute depth–age horizons.
The xsSO4 2– remaining after removal of the volcanic peaks is the robust-spline smoothed xsSO4 2–. This remaining xsSO4 2– (rxsSO4 2–) is assumed to be a mixture of total marine biogenic SO4 2– and stratospheric background SO4 2–. The stratospheric background SO4 2– is potentially composed of non-explosive volcanic SO4 2–, an admixture of sources that reside in polar stratospheric clouds, and continental SO4 2– sources such as anthropogenic emissions and dust.
The Antarctic Atmosphere and SO42– Aerosols
Throughout the year, Antarctic coastal and low-elevation areas are strongly influenced by lower-tropospheric air masses compared to higher-elevation interior areas. The influence of mid-/upper-tropospheric and stratospheric air masses on coastal sites is minor (Reference MinikinMinikin and others, 1998; Reference Legrand and WagenbachLegrand and Wagenbach, 1999). The sea-salt fraction of the total SO4 2– budget is large at coastal and low-elevation sites, contributing over ∼25% to sites such as Siple Dome (Table 1). Several studies have shown that xsSO4 2– concentrations and deposition timing are similar from site to site around the Antarctic coastline (Reference Prospero, Savoie, Saltzman and LarsonProspero and others, 1991; Reference Legrand and PasteurLegrand and Pasteur, 1998; Reference MinikinMinikin and others, 1998; Reference Legrand and WagenbachLegrand and Wagenbach, 1999). This similarity implies that xsSO4 2– concentrations at coastal Antarctic sites are controlled by large-scale processes related to both the distribution of sulfur sources in the Southern Ocean and the atmospheric mixing and transport patterns (Reference Rankin, Wol and MartinProspero and others, 1991). Coastal xsSO4 2– concentrations are influenced throughout the year by long-range transport of marine biogenic emissions from mid-latitude sources located at 50– 608 S. During the summer months these same coastal sites are inundated by xsSO4 2– from marine biogenic sources located south of 60˚ S (Reference MinikinMinikin and others, 1998). Concentrations of xsSO4 2– at coastal sites exhibit a well-defined peak from November to January. This summer xsSO4 2– peak corresponds to the break-up of the Antarctic sea ice south of 60˚ S and a subsequent enrichment of chlorophyll concentrations (Reference MinikinMinikin and others, 1998). The marine biogenic xsSO4 2– component accounts for roughly two-thirds of the winter and >90% of the summer coastal xsSO4 2– concentrations (Reference MinikinMinikin and others, 1998). The remaining percentage of winter and summer coastal xsSO4 2– comes from sources such as long-range transport from continental areas and sedimentation from the stratospheric reservoir.
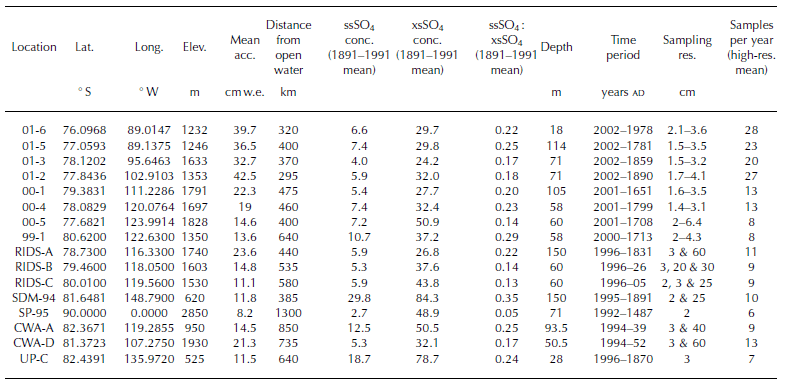
Table 1 Information for each ice core used in this study. Elev. = elevation; Mean acc. = mean annual accumulation; conc. = concentration; Sampling res. = sampling resolution; and Samples per year = mean number of samples per year calculated from entire high-resolution section of each core
High-elevation areas such as inland West Antarctica, South Pole and the Polar Plateau also receive SO4 2– from a variety of sources. The ssSO4 2– fraction peaks during the winter/spring transition months when intense cyclonic activity and intrusions of lower-tropospheric marine air masses are common (Reference Legrand, Feniet-Saigne, Saltzman and GermainLegrand and others, 1992; Reference Whitlow, Mayewski and DibbWhitlow and others, 1992; Reference HoganHogan, 1997). The oceans surrounding Antarctica are ice-covered during the winter/spring transition. Therefore, a low-/mid-latitude source is proposed for the ssSO4 2– (Reference Rankin, Wol and MartinProspero and others, 1991). By the time lower-tropospheric parcels of air reach the Polar Plateau they are severely depleted of heat, moisture and aerosols due to the long distances traveled (Reference Hogan, Barnard, Samson and WintersHogan and others, 1982; Reference PropositoProposito and others, 2002). As a result, ssSO4 2– accounts for <5% of the total SO4 2– budget on the Polar Plateau during the winter/spring and <1% during the summer (Reference Harder, Warren and CharlsonHarder and others, 2000; Reference Isaksson, Karlén, Mayewski, Twickler and WhitlowIsaksson and others, 2001).
Mid-/upper-tropospheric air masses carry xsSO4 2– aerosols emitted primarily from low-/mid-latitude marine biogenic productivity sources (Reference ShawShaw, 1982; Reference Legrand, Feniet-Saigne, Saltzman and GermainLegrand and others, 1992; Reference MinikinMinikin and others, 1998) as well as episodic xsSO4 2– input from volcanic eruptions (Reference Legrand, Feniet-Saigne, Saltzman and GermainLegrand and Wagenbach, 1999). The SO4 2– contained in these mid-/upper-tropospheric air masses dominates the summer SO4 2– budget of high-elevation Antarctic areas, such as inland West Antarctica, South Pole and the interior portions of the Polar Plateau (Reference Delmas and BoutronDelmas and Boutron, 1978; Reference DelmasDelmas, 1982; Reference Kreutz, Mayewski, Twickler and WhitlowKreutz and Mayewski, 1999; Reference Harder, Warren and CharlsonHarder and others, 2000; Reference Isaksson, Karlén, Mayewski, Twickler and WhitlowIsaksson and others, 2001).
High-elevation Antarctic areas are also impacted by the stratospheric SO4 2– layer. The stratospheric contribution of SO4 2- is generally assumed to be weak (Reference Legrand and MayewskiLegrand, 1997; Reference Bergin, Davidson, Dibb, Jaffrezo, Kuhns and PandisBergin and others, 1998) except after global-scale volcanic eruptions (Reference Legrand and DelmasLegrand and Delmas, 1987; Reference Dibb and WhitlowDibb and Whitlow, 1996). The SO4 2– aerosols in the stratosphere sink and mix downward into the troposphere. The slow sedimentation of SO4 2– from the stratosphere is periodically enhanced by disruptions to the tropopause that often occur during the spring break-up of the polar vortex (Reference Saxena, Anderson and LinSaxena and others, 1995).
Methodology
Of the sixteen cores used in this study, eight have been previously reported. The Siple Dome core (SDM94) and the three Ross ice drainage system cores (RIDS-A, -B and -C) were collected by a University of New Hampshire team in 1994 and 1995 respectively (Reference Mayewski, Twickler and WhitlowMayewski and others, 1995; Reference Kreutz, Mayewski, Twickler and WhitlowKreutz and others, 1996, 1999, 2000). The South Pole core (SP95) was collected by the Polar Ice Coring Office (PICO) in the designated clean-air sector, ∼1.5 km upwind from South Pole Station in 1995 (Reference BattleBattle and others, 1996; Reference Meyerson, Mayewski, Kreutz, Meeker, Whitlow and TwicklerMeyerson and others, 2002). The central West Antarctic cores (CWA-A and -D) were collected during the 1994/95 field season by a University of Wisconsin team, and the results are reported by Reference Reusch, Mayewski, Whitlow, Pittalwala and TwicklerReusch and others, (1999). The Up-C ice core was collected during the 1995/96 field season by PICO for Pennsylvania State University.
The eight new cores used in this study were drilled during the US International Trans-Antarctic Scientific Expedition (ITASE) West Antarctic traverses of 1999–2001 and analyzed at sub-annual, continuous resolution (Table 1). The samples were melted using the University of Maine continuous melter system, yielding, for this study, an average resolution of ∼50 samples per meter. This high sampling resolution captures the clear seasonal signal that is present in each ion time series (Fig. 2). All samples were examined for their soluble major-ion content (Na+, K+, Mg2+, Ca2+, Cl–, NO3 –, SO4 2–). Each was analyzed using a Dionex® DX-500 ion chromatograph coupled to a Gilson® autosampler. To determine anion (Cl–, SO4 2– and NO3 –) concentrations, the chromatograph was set up with an AS-11 column with 6 mM NaOH eluent. For cation (Na+, Ca2+, Mg2+ and K+) concentrations, a CS-12a column with 25 mM MSA eluent was used. All ion concentrations are determined with an accuracy of better than 0.1 ppb.
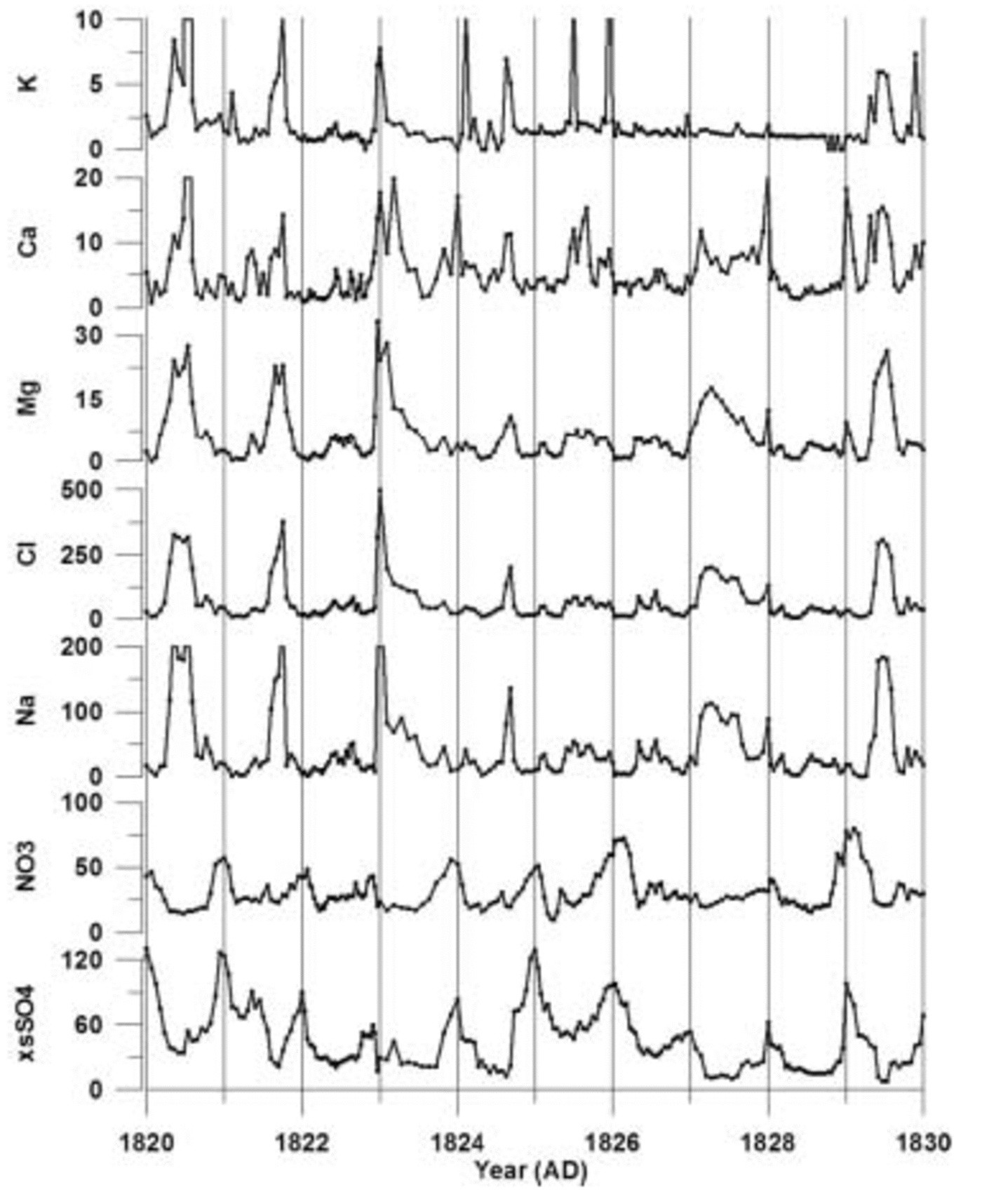
Fig. 2. Raw ion data from core 01-5 plotted vs time. Ion concentrations are in parts per billion. These data are from 94–99m depth (AD 1820–30) in the core. Vertical lines represent 1 year increments. is not constrained sufficiently for corrections to be made to our suite of cores.
The 1999–2001 cores are annually dated by matching seasonal peaks from each of the ion time series (Fig. 2). A ‘core-chemistry’ year is defined by a winter–spring peak in Na+, K+, Mg2+, Ca2+ and Cl– combined with spring–summer peaks in both NO3 – and xsSO4 2–, in accord with the seasonal timing identified by previous research (e.g. Reference Whitlow, Mayewski and DibbWhitlow and others, 1992; Reference Wagenbach, Wolff and BalesWagenbach, 1996; Reference Legrand and MayewskiLegrand and Mayewski, 1997; Reference Junge, Chagnon and MansonKreutz and Mayewski, 1999). Counting the seasonal layers down to absolute depth–age horizons (e.g. the Tambora 1815 volcanic event) demonstrates that each year is preserved in the sub-annual records of each core (Fig. 3) and that lower-resolution records cannot be used to accurately date volcanic events. For this reason, only the high-resolution portion of each ice-core record is used in this study. Based on several well-documented global-scale volcanic depth–age horizons, a dating accuracy of better than 1 year between known volcanic events is achieved for each SO4 2– record. The ssSO4 2– fraction is calculated by applying a standard sea-water ratio of 30.61 (Na+), 1.1 (K+), 3.69 (Mg2+), 1.16 (Ca2+), 55.04 (Cl–) and 7.68 (SO4 2–) to the ion concentrations in each sample (Reference HollandHolland, 1978). The concentration values are reduced incrementally according to this ratio until a value of zero is reached in one of the six ion concentrations. The ion which reaches zero concentration first is considered to be the conservative ion for that sample, and the concentration values for the other five ions are recorded. These become the excess (xs) concentrations for that sample. This technique, from O’Brien and others (1995), resulted in Na+ being the conservative ion for >90% of the samples in each core in this study. Recent studies have noted the effect of frost flowers on the ss fraction of ice-core chemistry. Frost flowers are depleted in SO4 2– relative to Na+ and this produces an ssSO4 2– value which is slightly higher than it should be for sites near the coast (Reference Reusch, Mayewski, Whitlow, Pittalwala and TwicklerRankin and others, 2002). At this time, the magnitude of the fractionation effect
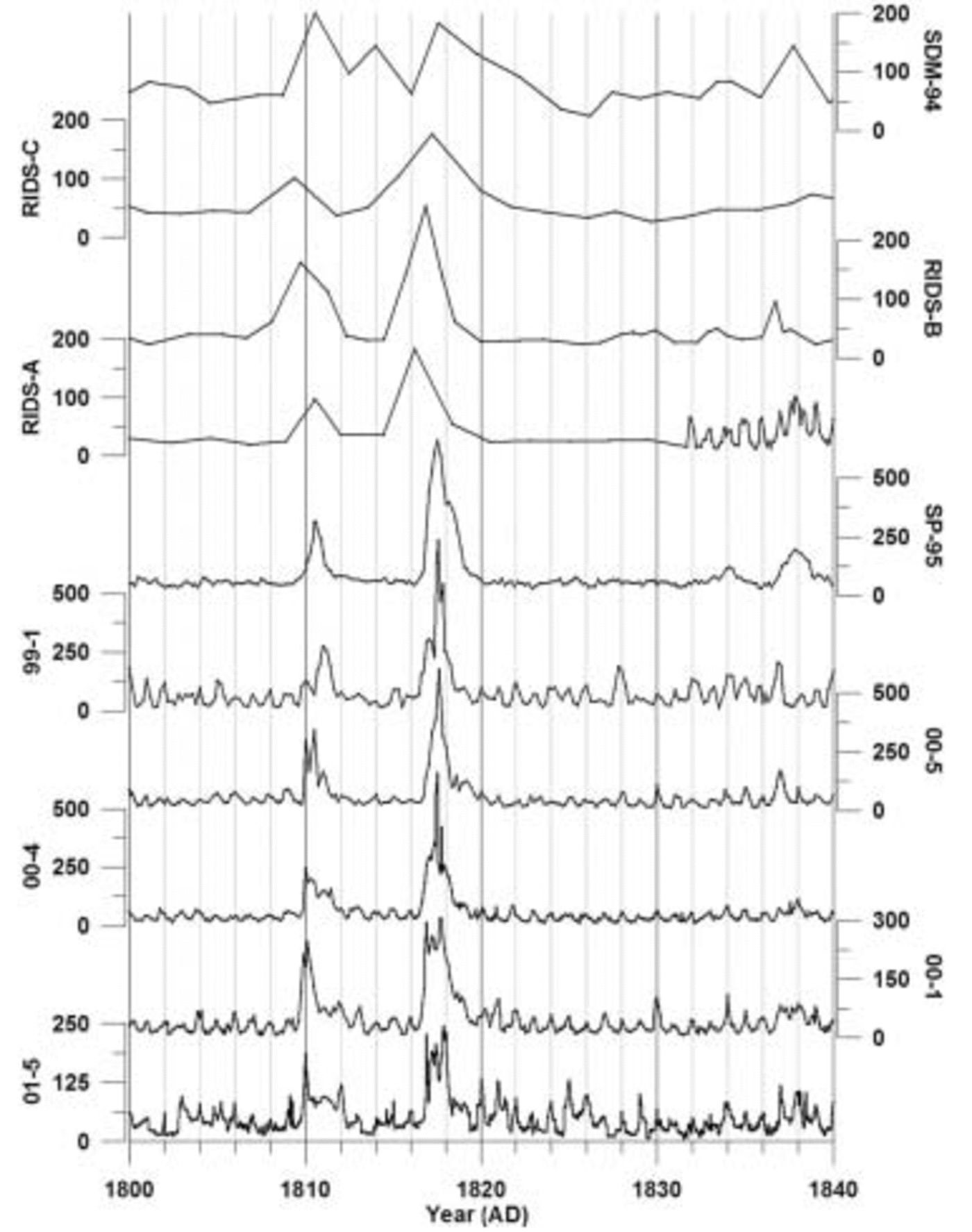
Fig. 3. Sub-annual and biannual raw excess sulfate ion concentration data for the years 1800–40 for ice cores 01-5, 00-1, 00-4, 00-5, 99-1, SP-95, RIDS-A, RIDS-B, RIDS-C and SDM-94. All concentrations are in parts per billion. Vertical lines represent 2 year increments.
In order to examine the spatial pattern of ss and xsSO4 2–, mean concentrations from 1891 to 1991 (apart from core 01-6 which only has a 23 year record) are plotted vs site elevation, straight-line distance from the nearest open water, and mean annual accumulation rate from 1891 to 1991 (Fig. 4). The reason for averaging over this time period is that any variability at lesser time-scales will be averaged out. Similar plots were constructed using ss and xsSO4 2– concentrations averaged from 1891 to 1940 and 1940 to 1991 to see if mean values from these different time periods affected the results. The values, although slightly different, retained the same trends with respect to elevation, straight-line distance from the nearest open water and mean annual accumulation rate. The temporal pattern of ssSO4 2– is examined by plotting the mean annual concentrations vs time for each core (Fig. 5). A 5 year running average is superimposed over these annual data to highlight any long-term trends. Empirical orthogonal function (EOF) analysis is used on the annual ssSO4 2– and xsSO4 2– concentrations for the years 1891–1991 and 1939–91 to see if any site-to-site associations exist in the ice-core records. These two time periods are used for the EOF calculations because the former represents the longest period of overlap between 10 of the ice-core records and the latter represents the longest period of overlap between 13 of the records (the record from 99-1 was not used in the EOF analysis because of data gaps).
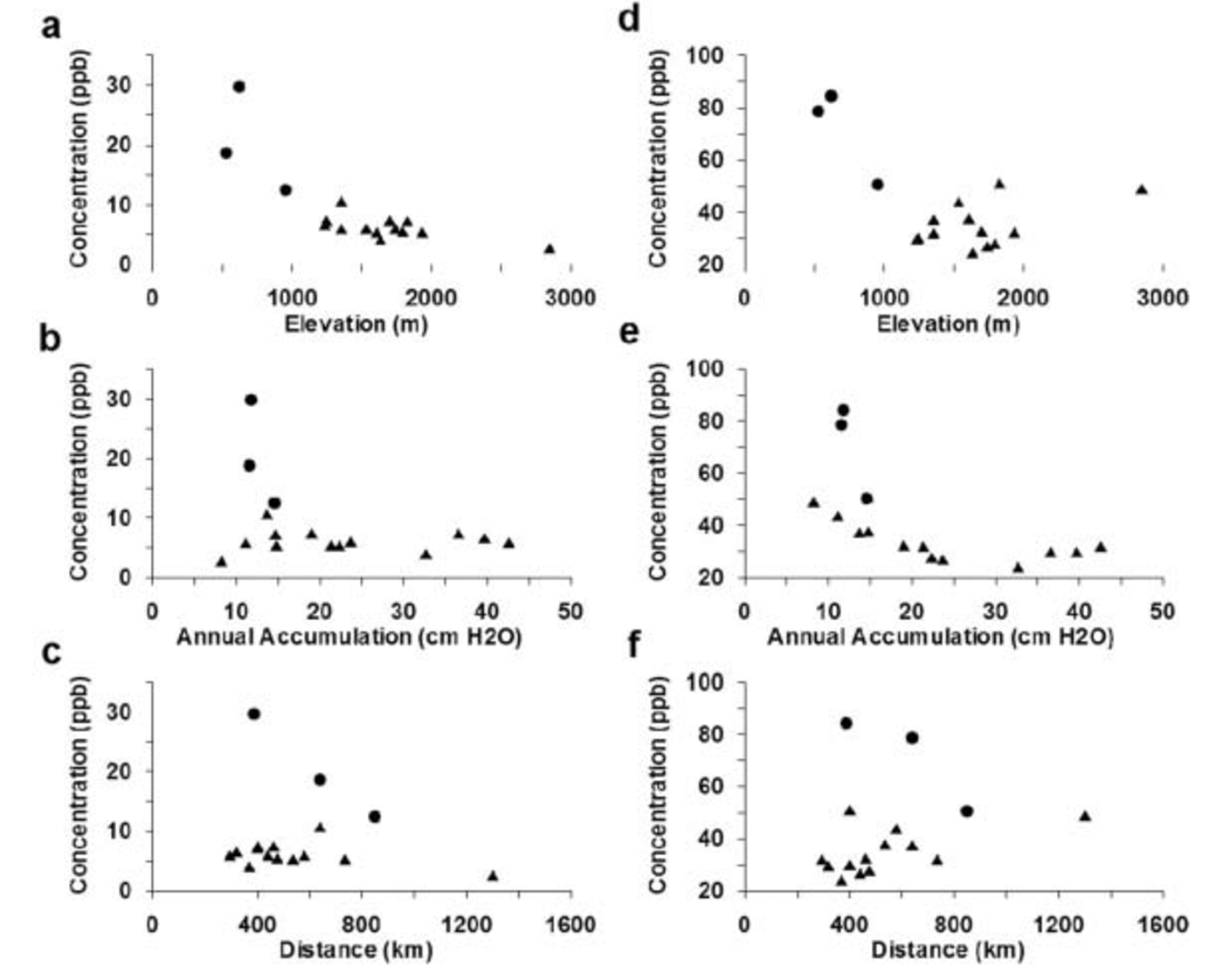
Fig. 4. Mean annual accumulations, ssSO4 2– and xsSO4 2– concentrations for the years 1891–1991. (a) ssSO4 2– vs elevation; (b) ssSO4 2– vs mean annual accumulation; (c) ssSO4 2– vs straight-line distance from nearest open water; (d) xsSO4 2– vs elevation; (e) xsSO4 2– vs mean annual accumulation; and (f) xsSO4 2– vs straight-line distance from nearest open water. Distances to open water are ignoring sea ice. All concentrations in parts per billion.
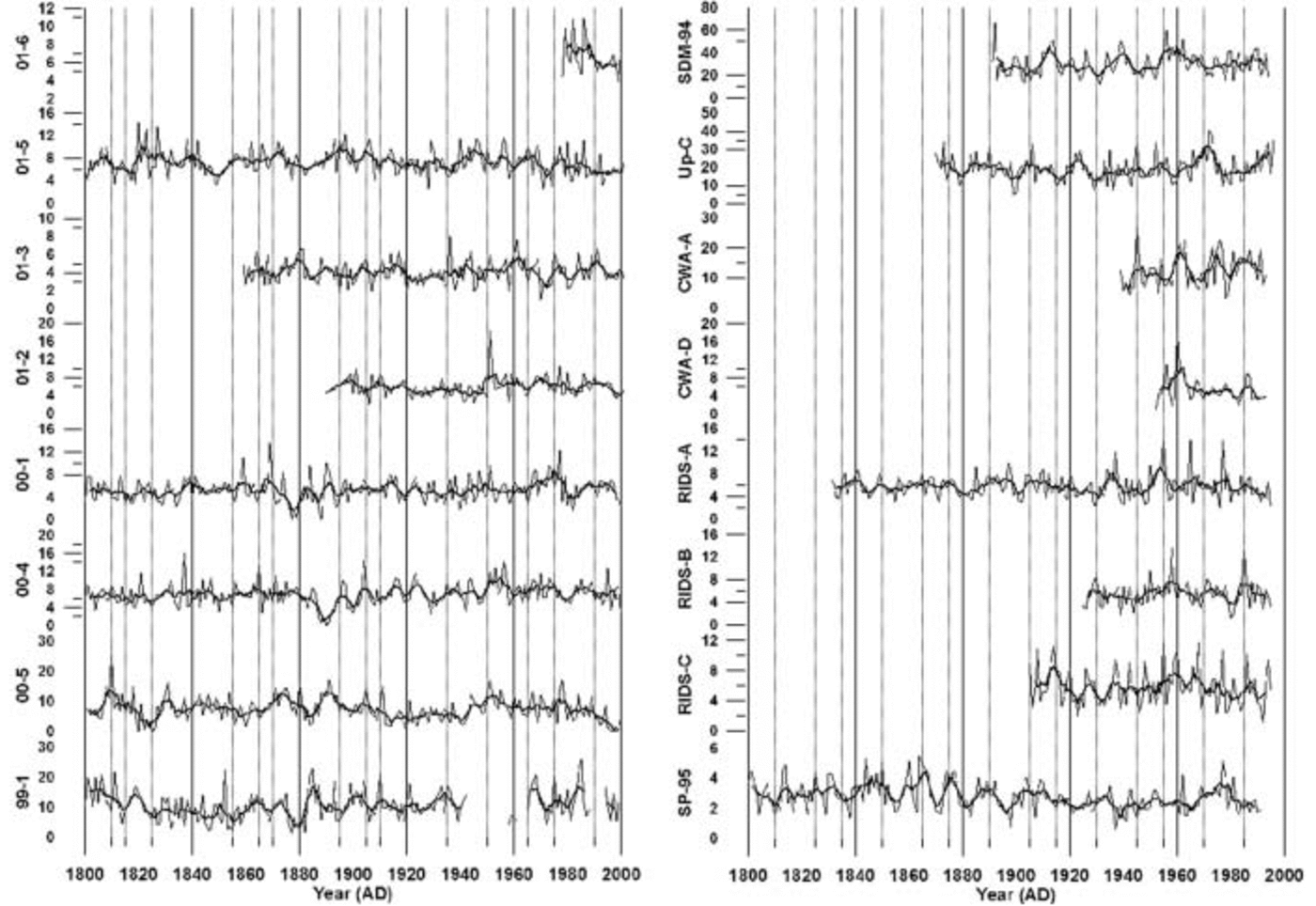
Fig. 5. Annual sea-salt sulfate concentrations in parts per billion for the years 1800–2002 for each ice core used in this study. Lighter lines represent annual concentration values, and thick darker lines represent a 5 year running average. Vertical lines represent 5 year increments.
Volcanic peaks are first identified from the xsSO4 2– data using a robust spline. A robust spline has the advantage over most smoothing functions that it is not affected by large outliers (e.g. volcanic peaks). The robust-spline tool outputs two separate time series which, when combined, make up the original series. One of these time series is the robust-spline smoothed series and the other is the robust-spline residuals (peaks). The amount of smoothing applied to each xsSO4 2– series is 80–90%, as this value provides the best approximation of the mean non-volcanic background concentrations beneath large volcanic spikes compared to adjacent years without volcanic SO4 2–.
The first step of the robust-spline technique is to average the raw xsSO4 2– data to annual resolution. The annual average calculation incorporates the raw background xsSO4 2– concentrations into the annual values. Thus, a large volcanic event that manifests as a significant rise in background xsSO4 2– concentrations will be recognized by the robust spline. The robust-spline function is applied to the annually averaged xsSO4 2– time series and the robust-spline smoothed series, and associated xsSO4 2– residuals are extracted. The mean residual xsSO4 2– concentration is calculated from the positive post-1825 xsSO4 2– residual values (this prevents the large Tambora volcanic xsSO4 2– signal from causing an anomalously high mean residual xsSO4 2– value). The mean +1σ and mean +2σ residual xsSO4 2– values are used to isolate the larger peaks from the xsSO4 2– residuals. Each residual xsSO4 2– peak greater than the mean +2σ value represents a strong volcanic signal, and each residual xsSO4 2– peak greater than the mean +1σ value represents a probable volcanic signal (hereafter referred to as 1σ and 2σ peaks). The 1σ and 2σ peaks are plotted on the same time axis for each core (Fig. 6).
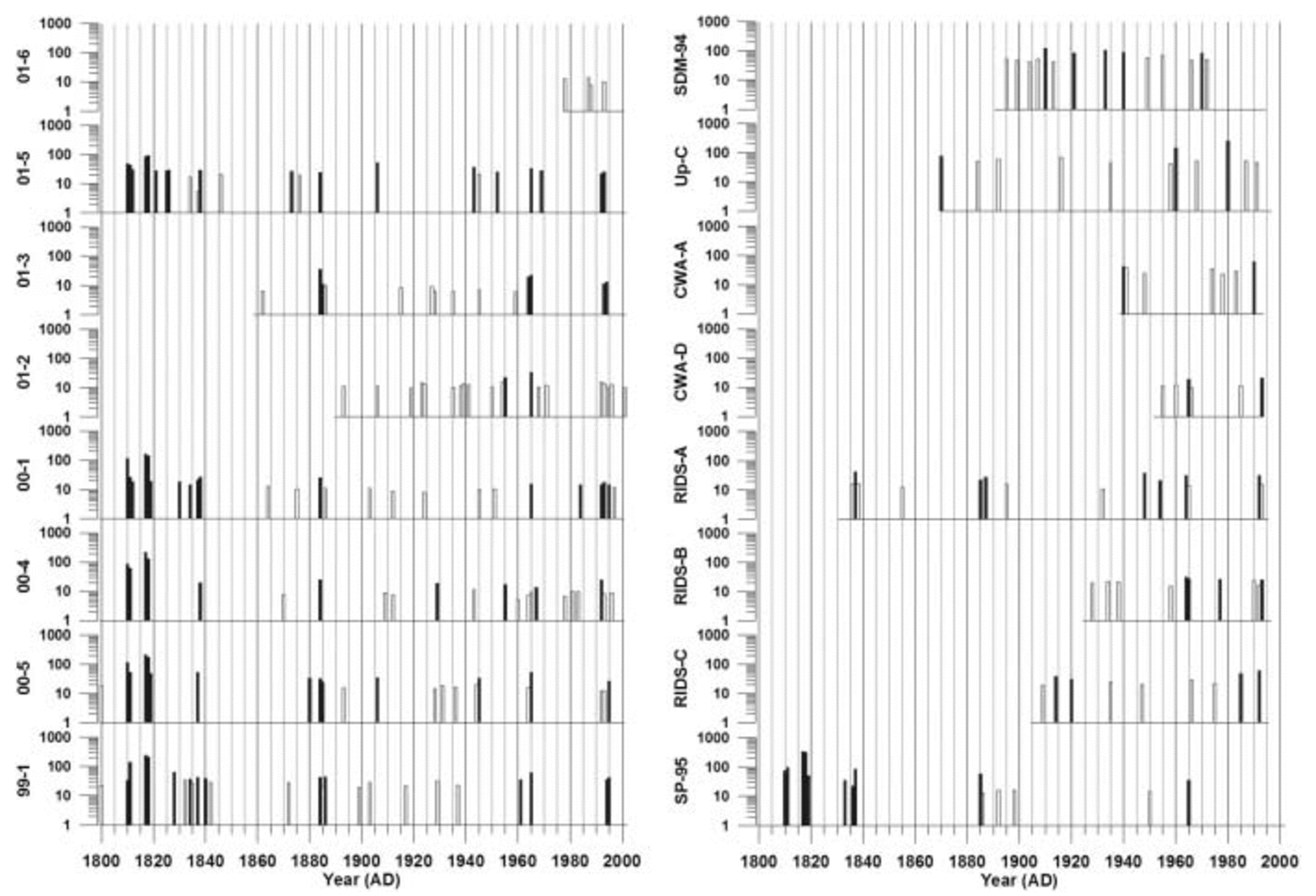
Fig. 6. Positive residuals from robust-spline smoothed annual excess sulfate concentrations for each ice core for the years 1800–2002. Peaks that are mean +1σ above the mean are shown in white, and peaks that are mean +2σ above the mean are shown in black. All concentrations are in parts per billion. Vertical lines represent 5 year increments.
As it is not currently possible to unambiguously separate the total marine biogenic and background stratospheric rxsSO4 2– components, the rxsSO4 2– time series for each core is used to examine the long-term trends in these two components over the last 200 years (Fig. 7). EOF analysis is used to determine if any associations exist between the rxsSO4 2– records from 1891 to 1991 and from 1939 to 1991.
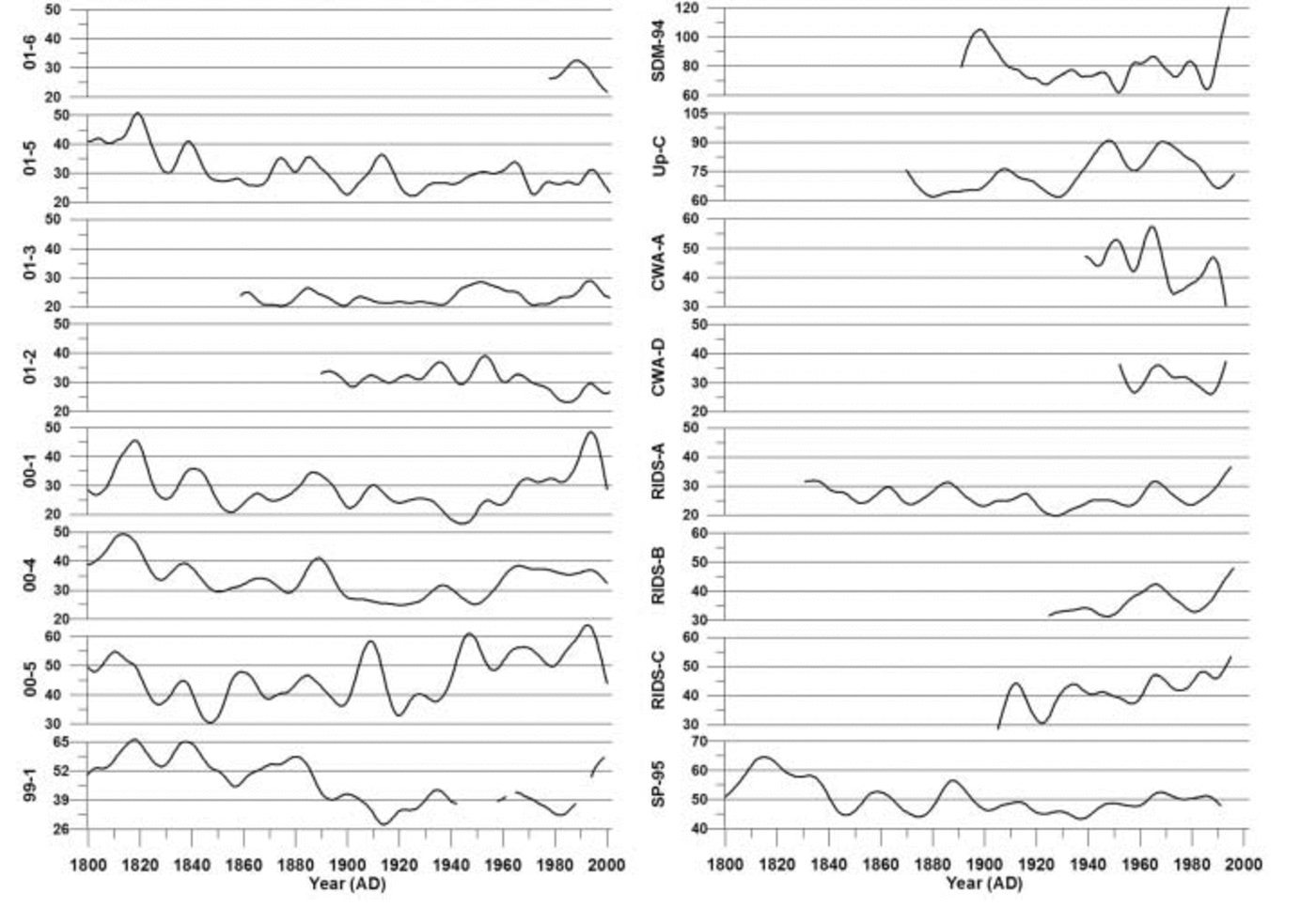
Fig. 7. Robust-spline smoothed annual excess sulfate (rxsSO4 2–) concentrations for each ice core for the years 1800–2002. All concentrations are in parts per billion. Vertical lines represent 10 year increments. Note scale change from site to site.
Results and Discussion
Ion concentrations and accumulation rate
Previous research reveals no significant correlations between snow ion concentration and accumulation rate (e.g. Reference Mulvaney and WolffMulvaney and Wolff, 1994; Reference Kreutz, Mayewski, Twickler and WhitlowKreutz and Mayewski, 1999; Reference Kreutz, Mayewski, Meeker, Twickler and WhitlowKreutz and others, 2000). This suggests that any dilution effects which do exist are offset by additional inputs such as dry and fog deposition (Reference Bergin, Meyerson, Dibb and MayewskiBergin and others, 1995; Reference Kreutz, Mayewski, Meeker, Twickler and WhitlowKreutz and others, 2000). Varying wet-deposition scavenging ratios can further confuse these effects. EOF analyses of the mean annual ion concentrations and mean annual accumulations for each US ITASE core resulted in weak correlations for most cores. The mean annual values of accumulation were used because this was the best resolution achievable for accumulation rate. The correlations between annual accumulation rate and SO4 2– are inconsistent and highly variable from site to site. The associations range from almost zero to <50%. These results suggest that although some portion of the SO4 2– is being deposited by wet deposition, another portion is deposited without being associated with wet precipitation. Because the dry-deposition velocity and wet-scavenging ratio for SO4 2– in West Antarctica are not well constrained and there is no consistent association between ion concentration and accumulation rate, flux corrections for accumulation were not applied.
Sea-salt SO42–
In Figure 4a the mean ssSO4 2– concentrations for sites located above 1000 m elevation (Fig. 1) are seen to display a relatively consistent range of values (∼4–7 ppb), apart from site 99-1 which has a mean ssSO4 2– concentration of ∼11 ppb. The high mean ssSO4 2– value observed at site 99-1 may be due to enhanced lower-tropospheric circulation near this site resulting in stronger advection of marine air masses to the area located over and to the east of the Ross Ice Shelf (Reference Kreutz, Mayewski, Twickler and WhitlowKreutz and Mayewski, 1999). Sites located below 1000m elevation (SDM-94, Up-C and CWA-A) have mean ssSO4 2– concentrations of ∼12–30 ppb and they are located to the west of site 99-1. The South Pole site has a very low mean ssSO4 2– concentration of ∼3 ppb.
Comparison between concentrations of ssSO4 2–, annual accumulation and distance from the coast (Fig. 4b and c) does not immediately reveal any clear trends, but if the sites below 1000m elevation, site 99-1 and the South Pole site, are ignored, the remaining sites display relatively similar ssSO4 2– concentration values (∼4–7 ppb). These results suggest that two distinct depositional regimes occur over West Antarctica. One affects sites above 1000 m elevation, and the other affects sites below 1000 m elevation. This supports the previous findings of Reference Kreutz, Mayewski, Twickler and WhitlowKreutz and Mayewski (1999) that lower-tropospheric circulation is enhanced in the SDM-94 vicinity because of stronger advection of marine air masses to this area. The most likely aerosol source for the low-elevation sites to the east of the Ross Ice Shelf is the Ross Sea.
Inspection of the annual ssSO4 2– concentrations reveals no significant long-term trends (Fig. 5). However, sites with the highest concentrations display the greatest concentration variability. An EOF (Table 2a) performed on ten cores with annual ssSO4 2– values spanning the years 1891–1991 reveals a relatively strong association (EOF1, 20% total variance and 21–46% variance of each core) of SP-95 with other sites located above 1000 m elevation apart from site 01-5 (Table 2a). This suggests that South Pole receives a significant fraction of its ssSO4 2– aerosols from the same air masses that supply West Antarctic sites located above 1000 m elevation. Statistical associations also occur between sites 01-5 and SDM-94: for example, in Table 2a EOF4 (10% total variance, 32% and 44% variance of each core) these two sites are positively correlated, and in Table 2a EOF6 (9% total variance, 40% and 23% variance of each core) they are negatively correlated. This positive– negative association may be related to findings by Reference Bromwich, Monaghan and GuoBromwich and others (2004) who noted that the El Niño–Southern Oscillation (ENSO) phenomenon causes a pronounced dipole structure over the Ross Ice Shelf–Marie Byrd Land area and over the Weddell Sea–Filchner/Ronne Ice Shelf. The dipole structure is observed in surface temperatures, meridional winds, cloud fraction and precipitation. An EOF of annual ssSO4 2– concentrations from 13 cores spanning the years 1939–91 yielded similar results (Table 2b) between SP-95 and other sites located above 1000 m elevation (EOF1, 19% total variance and 12–55% variance of each core). SDM-94 also displays similar positive–negative associations with site 01-5 (EOF2, 12% total variance, 37% and 16% variance of each core; EOF9, 6% total variance, 18% and 30% variance of each core). The similarity between these two EOFs implies that the ssSO4 2– deposition has remained relatively constant over West Antarctica for at least the last century.
Table 2 EOF of annual sea-salt sulfate concentrations for (a) 1891–1991 and (b) 1939–91. The row immediately below the EOF number row signifies total variance (%)

Excess SO42–
The xsSO4 2– signals for the ice-core records in this study are more complex than ssSO4 2– signals because the former have multiple sources. The location of a particular site with respect to physical parameters such as elevation and distance inland is critical for deciphering the xsSO4 2– signal. Examination of the data in Figure 4d reveals that xsSO4 2– concentrations decrease as elevation increases to approximately 1700 ma.s.l. Above this elevation, the decreasing trend in xsSO4 2– concentration switches and, as elevation increases, xsSO4 2– concentrations rise, although not as sharply as they decrease from sea level. This relationship illustrates the effects of multiple xsSO4 2– sources, local biogenic SO4 2– near the coast, and mid-/upper-tropospheric/stratospheric SO4 2– input to higher-elevation areas.
Two xsSO4 2– EOFs (Table 3a and b) demonstrate the separation between sites located above and below 1000m elevation (both EOF1, 29% total variance and 16–69% variance of each core located above 1000 m, 0–5% for cores located below 1000 m), suggestive of two separate source regions or transport pathways supplying xsSO4 2– to West Antarctica. All the core sites located above 1000 m elevation exhibit a decrease in xsSO4 2– concentrations as accumulation increases (Fig. 4e). However, this only occurs up to a threshold annual accumulation value of 20– 24 cm w.e. Sites with annual accumulation values greater than this threshold do not display any significant trends in xsSO4 2– concentration. Sites located below 1000 m elevation (SDM-94, Up-C and CWA-A) do not show any clear trends associated with annual accumulation, although these sites do exhibit a decreasing trend in xsSO4 2– concentrations vs distance from the nearest open water (Fig. 4f). Sites located above 1000 m elevation display the opposite trend: increasing xsSO4 2– concentrations vs distance from the nearest open water (Fig. 4f). These results suggest that sites located above 1000 m elevation receive the majority of their xsSO4 2– from mid-/upper-tropospheric/stratospheric air masses and that sites located below 1000 m elevation do not receive a significant fraction of their xsSO4 2– from this source. Previous work suggests that the xsSO4 2– supplied to sites located below 1000 m is deposited from lower-tropospheric air masses (Reference KreutzKreutz and Mayewski, 1999).
Table 3 EOF of annual excess sulfate concentrations for (a) 1891–1991 and (b) 1939–91. The row immediately below the EOF number row signifies total variance (%)
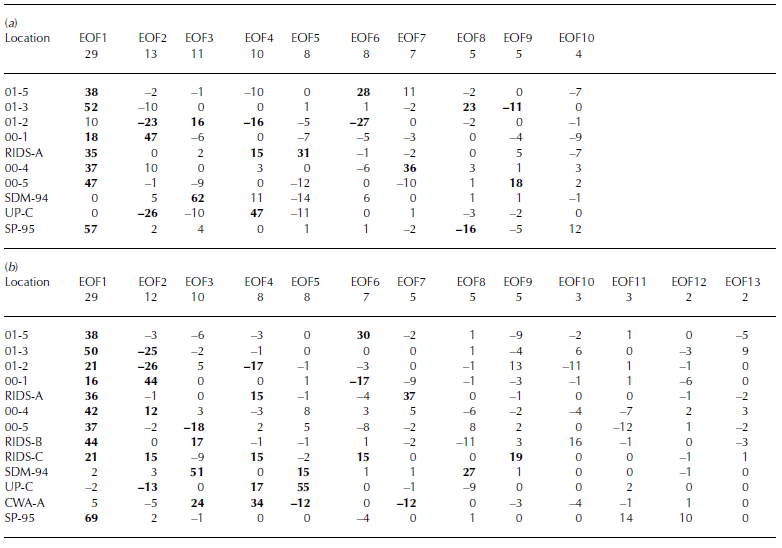
Remaining excess SO42–
Examination of the rxsSO4 2– curves in Figure 7 reveals that an overall decreasing trend takes place in all records from 1800 to ∼1940. Several of the rxsSO4 2– records exhibit an increasing trend from ∼1940 to ∼1990 (00-1, 00-4, 00-5, RIDS-A, RIDS-B, RIDS-C and, to a lesser extent, SP-95). Several sites exhibit a decrease in rxsSO4 2– during this same time period (01-5, 01-3 and 01-2). The remaining records do not show any significant trends or are too short to make an informed determination (site 99-1 is ignored beyond ∼1940 because of missing data). The opposing rxsSO4 2– trends observed in our records suggest that the source is not anthropogenic. If an anthropogenic rise in rxsSO4 2– were occurring, it should occur in all rxsSO4 2– records simultaneously. A second argument against an anthropogenically forced rise in rxsSO4 2– is the fact that the rise which does occur from 1939 to 1991 does not increase rxsSO4 2– levels higher than they were during the early 19th century.
EOF results of rxsSO4 2– for the years 1891–1991 (Table 4a) reveal a strong association (EOF1, 40% total variance and 13–77% variance of each core) between cores 01-5, 01-3, 00-1, RIDS-A, 00-4, 00-5, Up-C and SP-95. This 1891–1991 EOF captures the significant structure in the rxsSO4 2– curves. The majority of this structure consists of broad rxsSO4 2– peaks centered around ∼1910, ∼1947 and 1965 (Fig. 7). The large 1965 rxsSO4 2– peak indicates that some fraction of volcanic SO4 2– is still present in these records after the robust-spline residuals are removed. However, the ∼1910 and ∼1947 rxsSO4 2– peaks may be a result of local volcanic eruptions or increases from non-volcanic sources such as marine biogenic SO4 2–.
Table 4 EOF of robust-spline smoothed annual excess sulfate concentrations for (a) 1891–1991 and (b) 1939–91. The row immediately below the EOF number row signifies total variance (%)
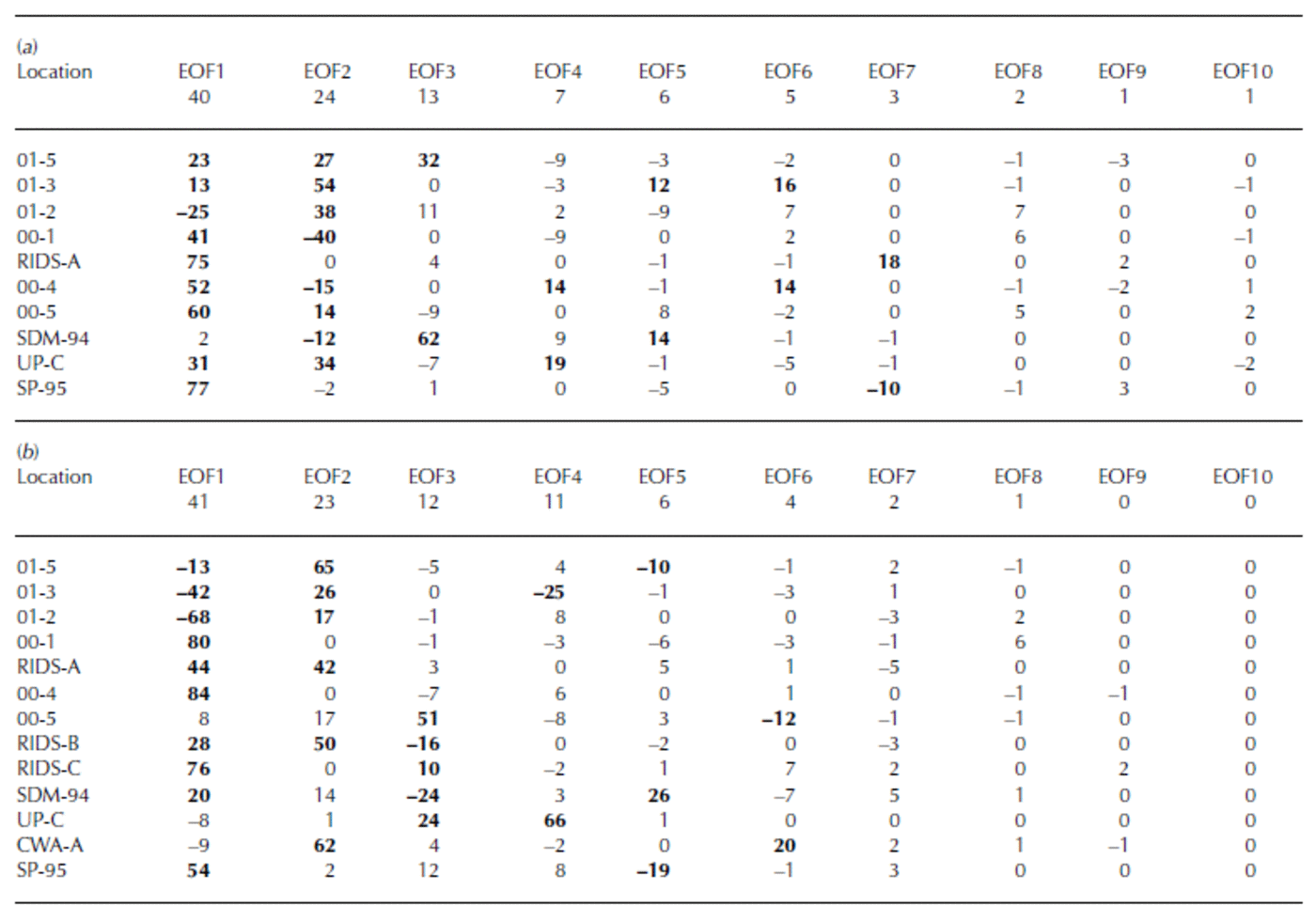
The EOF results of rxsSO4 2– for the years 1939–91 reveal a strong negative association (EOF1, 41% total variance and 13–84% variance of each core) between two groups of cores (Table 4b). The first group consists of 01-5, 01-3 and 01-2. The second group consists of 00-1, 00-4, RIDS-A, RIDS-B, RIDS-C, SDM-94 and SP-95. This 1939–91 EOF captures the overall long-term trend in rxsSO4 2– that takes place during this time period. The reason for the negative association is that the first group of cores (01-5, 01-3 and 01-2) display a decreasing trend in rxsSO4 2– concentrations from 1939 to 1991 and the second group (00-1, 00-4, RIDS-A, RIDS-B, RIDS-C, SDM-94 and SP-95) displays an increasing trend (Fig. 7). The trends in these rxsSO4 2– records suggest that a climate transition takes place over West Antarctica during ∼1940. At this time the atmospheric circulation over West Antarctica changes in such a way as to increase the transport of rxsSO4 2– to central West Antarctica.
Volcanic SO42– peaks
There are many volcanic events evident in the 16 ice-core xsSO4 2– records used in this study (Fig. 6). The strength of the volcanic signal preserved in polar ice varies according to the location of the core and the size, sulfur content, explosivity and location of the eruption. Global-scale volcanic eruptions, such as Pinatubo 1991, erupt with sufficient explosive force to inject large amounts of volcanic material directly into the stratosphere. Significant amounts of H2S and SO2gas are emitted during a global-scale eruption. The H2S is quickly converted to SO2, and this volcanic SO2 is subsequently oxidized to H2SO4 (Reference Junge, Chagnon and MansonJunge and others, 1961; Reference Berresheim, Wine, Davis and Singh, H.B.Berresheim and others, 1995). Elevated stratospheric SO4 2– concentrations can take as long as 3–4 years to return to pre-volcanic levels following a global-scale eruption (Reference Self, Rampino and BarberaSelf and others, 1981). As a result, volcanic SO4 2– in the stratosphere can spread over the entire globe before it is removed by mechanisms such as sedimentation.
In this study, global-scale eruptions (e.g. Tambora 1815, Krakatau 1883 and Agung 1963) appear simultaneously as 2σ peaks in all of the robust-spline residual xsSO4 2– records from sites located above 1000m elevation, but they are not obvious at sites located below 1000m elevation, such as SDM-94, Up-C and CWA-A (Fig. 6). The lack of global-scale volcanic eruption signatures in low-elevation records supports the idea that coastal and low-elevation sites in Antarctica are not strongly influenced by mid-/upper-tropospheric/stratospheric air masses.
Conclusions
The 16 high-resolution sub-annual records presented in this study preserve every year of chemical deposition in detail for the full length of each record. The dating accuracy of these records is estimated to be better than 1 year. Each record is calibrated using several known global-scale volcanic horizons. The detailed preservation of annual layers in the ice cores provides a remarkable opportunity for the investigation of major contributions to the Antarctic SO4 2– budget, notably: ssSO4 2–, volcanic-event xsSO4 2– and rxsSO4 2–.
These data show that sites located below 1000 m elevation (SDM-94, Up-C and CWA-A) receive higher concentrations of ssSO4 2– than sites located above 1000 m elevation. EOF results suggest that the sites located above 1000 m elevation receive a large percentage of ssSO4 2– from common air masses/sources, and sites located below 1000 m elevation receive ssSO4 2– from common air masses/sources that are separate from the air masses/sources supplying sites located above 1000 m elevation.
Concentration trends imply a dual source for West Antarctic xsSO4 2–: lower-tropospheric near the coast and mid-/upper-tropospheric/stratospheric for higher-elevation areas. These results highlight the relative importance of different xsSO4 2– sources and pathways for sites at different elevations. The rxsSO4 2– records reveal an overall concentration decrease at all sites over the period 1800 to ∼1940. Several of the rxsSO4 2– records also exhibit an increasing trend from ∼1940 to ∼1990, and several exhibit a decrease during this same time period. These rxsSO4 2– trends suggest that a significant shift in the spatial distribution of SO4 2– took place over West Antarctica at ∼1940. The atmospheric circulation changed in such a way as to increase the transport of rxsSO4 2– to inland West Antarctica and decrease it to other West Antarctic sites. This implies a major change in atmospheric circulation at this time which could not have been inferred from a single ice core, highlighting the importance and value of having an array of accurately dated spatially distributed records. Our records do not imply an anthropogenic source for any of the observed trends in rxsSO4 2–.
In our records all global-scale volcanic eruptions appear as 2σ peaks, as determined using residuals from a robust spline of the xsSO4 2– records. Global-scale volcanic eruptions cannot be consistently resolved in the xsSO4 2– records for sites located below 1000m elevation because the majority of volcanic SO4 2– from global-scale eruptions is transported to Antarctica through the mid-/upper-troposphere/stratosphere, and sites located below 1000 m elevation do not receive a significant fraction of xsSO4 2– from this source. High ssSO4 2– and local marine biogenic SO4 2– further obscure much of the volcanic SO4 2– present at these lower-elevation sites. The xsSO4 2– signals from global-scale volcanic eruptions can be used as reliable depth–age markers in West Antarctic cores located above 1000m elevation, but cannot be used definitively at sites located below 1000 m elevation.











