


Introduction
Understanding the clinical impact of genetic variants is one of the grand scientific challenges of the twenty-first century. A typical person has around 3 to 3.5 million variants, or little changes that distinguish the individual’s genome from an idealized human reference genome (Kohane et al. Reference Kohane, Hsing and Kong2012; US Dep’t of HHS/FDA 2014c). The role, if any, that most variants play in human health has not yet been discovered. As many as half a million of a person’s detected variants may be novel or at least rare (Kohane et al. Reference Kohane, Hsing and Kong2012), which means scientists may have had little opportunity to study the variants and infer their effects on health.
A 2014 study concluded that fewer than 200 variants were well enough understood at that time to merit further scrutiny for potential medical significance after a person undergoes genomic testing (Dewey et al. Reference Dewey, Grove and Pan2014). As for the rest of the variants that a test detects, their clinical validity is uncertain or unknown. Clinical validity “addresses whether there is a strong and well validated association between having a particular gene variant and having a particular health condition and whether knowing that a person has the gene variant offers meaningful insight into the person’s health or reproductive risks” (Secretary’s Advisory Committee on Genetic Testing 2000). Once a variant has an “established” clinical meaning, a test that detects that variant is said to have clinical validity for purposes of diagnosing the associated health condition (Fabsitz et al. Reference Fabsitz, McGuire and Sharp2010).
Modern genomic tests provide a practical way to identify the many genetic variants that people have, but interpreting the meaning of those variants depends on an external body of knowledge: cumulative discoveries about the associations between genotype (the specific gene variants a person has) and phenotype (physical characteristics in which those gene variants may have played a role). For most of the variants that tests can detect, the tests lack clinical validity because these associations are not yet known.
This chapter focuses on genomic tests, which differ both in technology and in scale from the traditional genetic tests of the past. Traditional genetic tests – including many still used today – typically focus on one or a few specific genes and are designed to detect a predetermined list of variants that were known in advance to be pathogenic – that is, variants already known to have a negative impact on health (US Dep’t of HHS/FDA 2014c). Polymerase chain reaction (PCR) and single-nucleotide polymorphism (SNP) arrays are examples of these older genetic-testing technologies. In contrast, genomic tests include next generation sequencing (NGS) assays that can detect any variant present in a specific group of genes; whole genome sequencing (WGS) tests that detect variants in virtually the entirety of a person’s genome; whole exome sequencing (WES) tests that detect variants in the roughly 1.5 percent of the genome that contains the person’s genes; and copy number variant arrays (Evans, Burke, and Jarvik Reference Evans, Burke and Jarvik2015).
The US Food and Drug Administration (FDA) notes that genomic tests can produce large volumes of information that may include novel variants never seen before and variants that, while previously seen in other people, have unknown clinical impact (US Dep’t of HHS/FDA 2014c). This state of affairs creates concern about the safety of patients, research subjects, and other consumers (together, “consumers”) who undergo genomic testing. The FDA has expressed concern that consumers may pursue irreversible medical treatments – such as prophylactic surgeries to mitigate a suspected susceptibility to cancer – based on findings that later prove to have lacked any clinical validity (US Dep’t of HHS/FDA 2014a). Late in 2014, the agency sought comments on two Draft Guidances that proposed to regulate laboratory developed tests (LDTs) as medical devices; doing so would reverse a long-standing policy of using FDA’s enforcement discretion to avoid regulating most LDTs (US Dep’t of HHS/FDA 2014a, 2014b). FDA ultimately chose not to finalize the Draft Guidances, but identified a set of unresolved issues likely to reemerge as focuses of FDA regulatory attention in the future.
The two Draft Guidances defined a test’s safety and effectiveness in terms of two parameters: analytic validity (whether the test accurately detects the variants it purports to detect) and clinical validity (whether the detected variants have a well-established association with specific clinical conditions that can be diagnosed or predicted based on the test results) (US Dep’t of HHS/FDA 2014a). It was apparent that FDA’s traditional premarket review process for medical devices was not well suited to the task of evaluating the safety and effectiveness of genomic tests: how can test sponsors be required to prove – in advance – the clinical validity of the many millions of genetic variants that a test may detect, when the full range of variants that exists in the human population only becomes known after the test is approved and moves into wide clinical use?
In response to this dilemma, FDA published a discussion paper proposing to rely on high-quality, curated genetic databases to establish the clinical validity of genomic tests (US Dep’t of HHS/FDA 2014c). This approach would draw on an external body of cumulative knowledge – all that is known about the genome, as reflected in well-curated, external databases at any given time – rather than requiring test manufacturers to generate from scratch the entire body of evidence needed to support FDA’s review of a new test. The agency convened a workshop in February 2015 to discuss this proposal and subsequently held two further public workshops in November 2015 to explore analytical performance standards for NGS tests and ways to harness databases to ensure their clinical validity (US Dep’t of HHS/FDA 2015c, 2015d).
As FDA and other policymakers continue to develop approaches for regulating genomic tests, one thing seems clear: to make genomic testing safe and effective for consumers, it will be necessary to develop large-scale genomic data resources. This chapter draws on the work of Elinor Ostrom to explore whether the human genome can be modeled as a common-pool resource (CPR). It concludes that this analogy is strong, at least at this time. The strength of the analogy may, however, be an artifact of the technologies – and the costs of those technologies – currently used to test the human genome. So long as the CPR analogy remains apt – and it may remain so for a period of years or even decades – lessons learned in managing other types of CPRs may shed light on how best to organize collective efforts to create and sustain genomic data resources and help maximize their productivity in terms of new discoveries and clinically useful insights that improve human health.
4.1 The Human Genome as a Natural Resource
Ostrom warned about the perils of using natural resource commons as an analogy for unrelated phenomena (Ostrom Reference Ostrom1990), yet the human genome is a natural resource in a very real sense. Like all other natural resources, the human genome is “found in nature and is necessary or useful to humans” (American Heritage Science Dictionary 2005). Naturally occurring gene sequences are not “made by man” but rather are “manifestations of … nature,” akin to a “new mineral discovered in the earth or a new plant found in the wild” (Diamond v. Chakrabarty, 447 US 303, 309 (1980); Funk Brothers Seed Co. v. Kalo Inoculant Co., 333 US 127, 130 (1948)). The Supreme Court’s 2013 decision in Association for Molecular Pathology (AMP) v. Myriad Genetics rightly characterized isolated gene sequences as non-patentable subject matter because they are products of nature (133 S.Ct. 2107 (2013)). Medical genetics can be modeled as an extractive industry that creates infrastructure – such as genomic testing instruments, laboratories, databases, and computer algorithms – to mine these natural resources and process the extracted raw commodity (information about the specific genetic variants detected in tested individuals) into useful products and services that improve human health (Evans Reference Evans2014a).
The fact that the human genome is a natural resource does not necessarily imply it is a CPR, as conceptualized in Ostrom’s work (Ostrom Reference Ostrom1990: 30). The relevant aspect of a CPR, for the present discussion, is that it is a resource system whose ultimate productivity can be optimized through coordinated action and sound governance. In the classic CPR context, the natural resources include fisheries, mineral deposits, or grazing areas – some renewable, some nonrenewable, depending on whether they can be replenished on a time scale relevant to humans. These resources are subject to congestion or depletion, and careful coordination is needed to manage those problems. In the genomic context, the challenge is somewhat different: coordination is necessary in order to create data infrastructures that make it possible to extract useful knowledge by testing people’s genomes. People have a vast supply of copies of their own genomes, which are replicated in every one of the cells of their bodies, but extracting useful information from this raw genetic material requires coordination with other people.
Ostrom defines a resource system as a stock that is “capable, under favorable conditions, of producing a maximum quantity of a flow variable without harming the stock or the resource system itself” (Ostrom Reference Ostrom1990: 30). As users appropriate “resource units” – for example, by harvesting fish from a fishery or withdrawing oil from a reservoir – their actions may affect the ultimate productivity of the resource system itself, for example, by collapsing the sustainability of a renewable system or by failing to optimize the total recovery from a nonrenewable one. When multiple individuals or firms jointly use a resource system, failure to coordinate their activities may diminish the ultimate flow of beneficial resource units for all. This aspect of CPR systems is highly relevant to genomics.
The unique genomes buried beneath the skins of human beings are a stock that can yield a flow of valuable resource units, but only in an environment of sustainable knowledge and data sharing. Individuals who undergo genomic testing – assuming each person acts in isolation and no data sharing occurs – would learn the configuration of nucleotides in their respective genomes, but they would have no basis to infer what this configuration implies about their present and future health. These latter inferences – in other words, useful knowledge about the clinical meaning of particular genetic variants – can only be made by pooling genetic and other types of health data for large samples of the human population and then drawing statistical inferences based on which variants and health conditions appear to occur together in the same individuals: for example, people with genetic variant X also tend to manifest disease Y, whereas the people without variant X seem not to develop disease Y as often.
The valuable resource units in genomics are discoveries establishing the clinical validity of specific gene variants, as well as clinically useful individual diagnoses based on those discoveries. Humankind’s collective stock of genomic material is a natural resource, but extracting value from it requires data infrastructure to study additional variants beyond those understood today. Each statistically valid inference that uncovers a new association between a specific gene variant and a specific health condition is one resource unit, in Ostrom’s terminology. The number and pace of these discoveries can be maximized under favorable conditions. What are those conditions, and can Ostrom’s work on other types of CPRs help identify them?
In principle, the genomic resource system that can be studied includes the genomes of all living people plus any deceased individuals for which genomic data (or biospecimens that could be tested to derive genomic data) still exist (Evans Reference Evans2014a). In practice, genomes become part of the useable resource system only when people undergo genomic testing. Testing is the act that causes a person’s variants to come to light, like gold nuggets uncovered in the sand, so that their possible clinical impacts can be explored. Most people living in the early twenty-first century will never undergo genomic testing. As of 2014, only about 228,000 human genomes had been fully or partly sequenced worldwide, mostly in research settings because genomic testing has only recently begun to move into wider clinical use (Regalado Reference Regalago2014). The Precision Medicine Initiative announced by President Obama early in 2015 envisions testing a 1-million-person cohort of volunteers (The White House 2015; Collins and Varmus Reference Collins and Varmus2015). One million is an impressive figure but still a mere 14 thousandths of 1 percent of the entire human population. Shirts et al. (Reference Shirts, Jacobson, Jarvik and Browning2014) note that many of the genetic variants detected during evaluations of familial cancer risk have uncertain clinical significance, and very large sample sizes – hundreds of thousands to millions of individuals – may be required to ascertain the impact of these variants.
Discovering the clinical significance of not-yet-understood gene variants will require a much larger data infrastructure than exists today. The requisite data resources would include genome sequencing data for very large samples of the human population along with detailed phenotypic information – that is, data reflecting their past and present health conditions, environmental exposures, and lifestyle habits (Evans, Burke, and Jarvik Reference Evans, Burke and Jarvik2015). These data resources must be large scale (in the sense of including data for many individuals) and deeply descriptive (in the sense of containing much or all of the available health-related information for each of them) (Evans Reference Evans2016a).
Public research budgets – that is, funds available from governmental agencies such as the National Institutes of Health and its counterparts in other nations – will not suffice to sequence the needed number of genomes. The genomic data resource system needs to capture not just the genomes tested under grants of NIH research funding (Evans Reference Evans2016a). It also needs to include genomes sequenced in clinical settings as part of regular health care financed by public and private payers such as Medicare and health insurers, as payers begin covering clinical genomic testing (Evans Reference Evans2016a). Also important – potentially, very important – are the direct-to-consumer genomic test results of financially capable, curious consumers who take matters into their own hands when payers fail to cover the genomic tests consumers need or desire. These respective data streams are under radically different legal and regulatory regimes: federal research regulations and privacy laws for data generated during research; federal privacy law and a web of state privacy and medical records laws for the clinical data; and corporate privacy policies, including click-through contractual agreements, and state laws that address access to data from direct-to-consumer personal health services (Evans Reference Evans2016a).
The resource units to be extracted from this resource system include both broadly generalizable knowledge such as discoveries about the clinical impact of a human gene variant, as well as context-specific insights that improve the well-being of individuals (e.g., a more accurate interpretation of one patient’s genomic test) (OECD 2015: 29). This distinction is expressed in US regulations as the difference between generalizable knowledge that has the potential to benefit many people in the future (“research”) versus patient-specific inferences drawn during clinical care (“treatment”) (see, e.g., 45 C.F.R. § 164.501). Extracting value from this resource system involves four basic steps: (1) conducting individual genomic tests to detect which variants each individual has; (2) linking each tested individual’s genomic data to detailed phenotypic information such as longitudinal health records from the person’s encounters with the health care system as well as available environmental and lifestyle data, such as data from wearable fitness sensing devices; (3) pooling these linked genomic and health records for large populations of individuals; and (4) searching for statistically significant associations between specific gene variants and specific health conditions.
In step 1, NGS tests are an efficient way to identify the range of genetic variants each individual carries. While the cost of gene sequencing has dropped significantly in recent years, the cost of NGS testing is still high enough that, at this time, a person whose genetic variants are detected by one laboratory is unlikely to repeat the testing at a different laboratory (Evans Reference Evans2014a). In this respect, genomic testing differs from blood typing and many other common diagnostic tests that are easily repeated whenever a different physician or researcher needs to know the same information. This fact may change over time as the cost of gene sequencing drops, but, for now, a laboratory that detects a person’s variants in effect captures the resulting information.
Individual genomic test results – or more precisely, opportunities to conduct genomic testing – are, in effect, a nonrenewable resource: production of a person’s gene variant data by one laboratory effectively precludes production of those same data by a different laboratory. A vast collective human gene pool consists of two unique genomes (one from mom, one from dad) for each member of the human species. When a laboratory tests an individual, it appropriates that person’s genomic information, which is unlikely ever to be sampled again. Thereafter, the person’s genomic information is unavailable for others to study unless shared beyond the initial laboratory.
This remains true even in clinical settings, where a physician orders genomic testing to guide a patient’s health care. Clinical laboratories typically report only a tiny fraction of the variants genomic tests identify back to the physician for inclusion in the patient’s medical record. Yet the testing generates a massive amount of underlying data that, once generated, could be reused, repurposed, copied, and/or shared at a relatively modest marginal cost. The marginal cost of data sharing is not, however, zero (Evans Reference Evans2014b). It includes various items that HIPAA characterizes as costs of “data preparation and transmittal” (42 U.S.C. § 17935(d)(2)(B); 45 C.F.R. § 164.502(a)(5)(ii)(B)(2)(ii)). Examples include the costs of paying skilled personnel and tying up capital (such as information systems and software) to locate data responsive to a particular request; to convert those data into a consistent, machine-readable format that allows information from one source to be compared with data from external sources; to comply with legal and regulatory requirements, such as review by an Institutional Review Board, that may be necessary to effect a lawful data transfer, and to convey the data securely to the requesting party (Evans Reference Evans2014b). The fact that the marginal costs of data preparation and transmittal are nonzero has important implications for the development of large-scale genomic data resources. It means that designing a workable revenue model is essential to the task of developing sustainable genomic data resources, as discussed further later, in Section 4.4.
As for the types of “underlying” data that may be on file at a genomic testing laboratory, the initial step of genome sequencing produces image files and base call (BCL) files. These files are so large that laboratories process them in real time and almost immediately discard them because of data storage constraints (Evans et al. Reference Evans, Dorschner, Burke and Jarvik2014). The follow-on data analysis generates, in order: (1) FASTQ files containing raw sequences for fragments of the person’s genome, along with quality scores bearing on how reliable the information is; (2) BAM (binary alignment/map) files that map these raw sequences onto the reference human genome; and (3) a VCF (variant call format) file that summarizes the individual’s sequence variants, sorted by their positions in the genome (Evans et al., Reference Evans, Dorschner, Burke and Jarvik2014). Only a handful of the variants in the VCF file will have a known relevance to the particular medical condition that caused the patient to undergo genomic testing. Laboratories typically interpret only the medically relevant variants for reporting to the physician and patient. The remaining variants – 3 million or so in a patient undergoing whole genome sequencing – often remain in the laboratory’s files. Clinical laboratories are subject to data retention requirements under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) (42 U.S.C. § 263a; 42 C.F.R. § 493.1105). There are no direct regulatory instructions on how to apply these requirements to the various files genomic testing generates, but recent studies recommend retaining a patient’s VCF file and – if storage permits – the BAM and FASTQ files (Gargis et al. Reference Gargis, Kalman and Berry2012; Rehm et al. Reference Rehm, Bale and Bayrak-Toydemir2013).
To extract resource units (discoveries) from these individual genomic data files, they would need to be linked to the person’s other health data (step 2 of the discovery process) and studied alongside similar records for many other individuals (steps 3 and 4). This second step – linking an individual’s data files that have been stored by multiple data holders – requires at least some identifying information to verify that the files being linked all relate to the same individual (Evans Reference Evans2011: 93–94). The need for identifiers potentially adds to the transaction cost of assembling genomic data resources because, under the existing US federal regulations discussed in the next section, the sharing of identifiers may trigger the need to procure individual informed consents and/or privacy authorizations before data holders can share data (see discussion infra later in Section 4.2). If an individual’s FASTQ, BAM, and VCF files stay siloed at the clinical laboratory that ran the test, their full scientific value cannot be harvested. Fragmentation of genomic and health care data resources creates dilemmas that threaten to impede or block genomic discovery if collective action fails.
4.2 The Data Access Challenge
The existence of stored genomic and clinical data offers a major opportunity to develop immensely valuable public infrastructure in the form of large-scale data resources to support new discoveries about the clinical significance of human gene variants. There are, however, a number of barriers and obstacles to overcome – most notably, collective action and governance dilemmas that bear a familial resemblance to CPR and other shared resource contexts.
At present, each patient’s data – any available genomic test results as well as other health records – are scattered among many data holders such as physicians, hospitals, laboratories, and insurers with which the person has done business. Recent efforts to promote interoperable electronic health records have not injected order or consistency. The President’s Council of Advisors on Science and Technology (P-CAST), surveying the situation in 2010, despaired that a standard data format would ever emerge: “Any attempt to create a national health IT ecosystem based on standardized record formats is doomed to failure … With so many vested interests behind each historical system of recording health data, achieving a natural consolidation around one record format … would be difficult, if not impossible” (P-CAST 2010: 39). Even if data holders shared a common data format that enabled them to communicate with one another, they often do not want to do so: a study funded by the National Human Genome Research Institute surveyed a diverse group of genomics industry insiders who ranked data holders’ resistance to data sharing as the most important but least tractable policy challenge (McGuire et al. Reference 100McGuire2016).
Health data sharing has been characterized as a tragedy of the anticommons (Hall Reference Hall2010; Rodwin Reference Rodwin2010). Rodwin adds that, in reality, there are two distinct tragedies: the first reflects the fragmentation of genomic and other health data resources at the level of institutional and commercial data holders, such as hospitals, laboratories, and research institutions. The second tragedy unfolds at the level of individual patients and consumers, who can block the sharing of their data under a web of privacy and human subject protection regulations that, in many instances, condition data access on individual consent (Rodwin Reference Rodwin2010: 606). This two-tiered tragedy of the anticommons is the challenge to be overcome.
Elsewhere, I have analyzed this problem using the analytical framework for governing knowledge commons outlined by Frischmann, Madison, and Strandburg (Reference Frischmann, Madison, Strandburg, Frischmann, Madison and Strandburg2014). Key points can be summarized as follows: there have been persistent calls for individual ownership of health data, but the legal reality is that neither the individual nor the data holder has exclusive ownership of stored health data; the two parties share control (Evans Reference Evans2016a: 661–64; OECD 2015). Their shared control creates four possible pathways for assembling large-scale data resources, as portrayed in Figure 4.1.
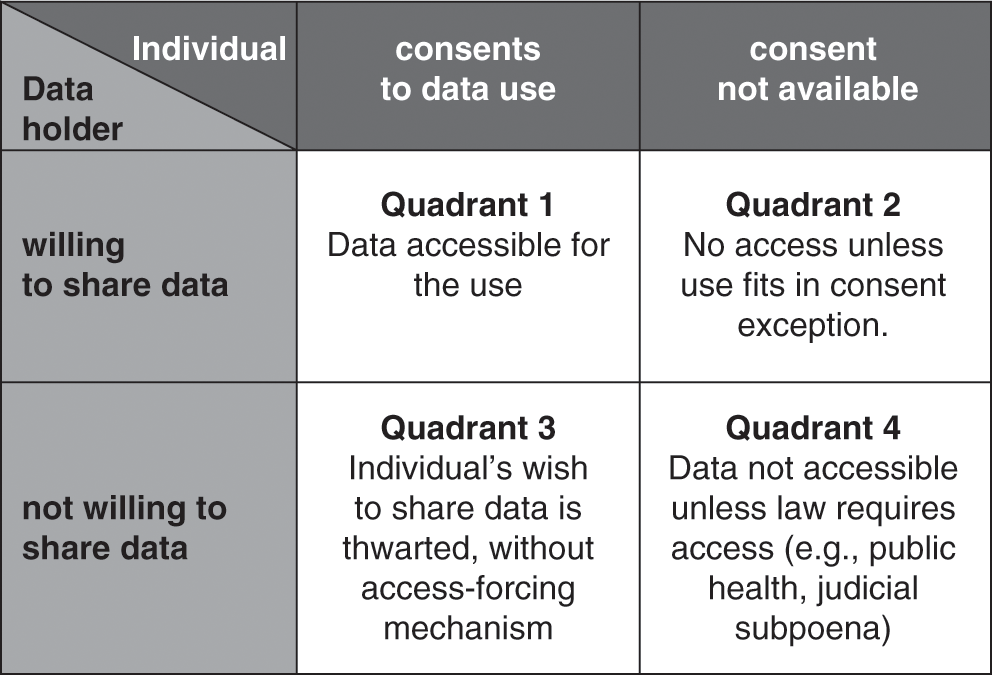
Figure 4.1 The challenge of access to commercially held clinical data (adapted from Evans Reference Evans2016a: 668, Figure 1).
In the best of all possible data-sharing worlds, there would be consent alignment, with the individual and the data holder both willing to share data (see Quadrant 1 in Figure 4.1). Both parties would facilitate flows of data to create a national genomic infrastructure. In theory, consent alignment could arise spontaneously or, perhaps, in response to educational stimuli such as a public information campaign highlighting the many benefits that large data resources could offer. To date, however, consent alignment has not emerged in our society at the level required to create multi-million-person genomic data resources. A variant of Quadrant 1 is incentivized consent alignment, in which a fiscally empowered entity – such as a government or a public research funding agency – uses conditional grants (offers of money that are contingent on the recipients’ agreement to share data). Related options would be to create tax incentives for data sharing or to condition a desired benefit (such as an FDA approval or a Medicare reimbursement) on data sharing (Evans Reference Evans2016a: 18).
The National Institutes of Health (NIH) and research funding agencies in other nations have made effective use of conditional spending and have fostered data sharing through policies that obligate grantee research organizations to deposit certain kinds of data generated using grant funds into shared databases (Contreras Reference Contreras, Frischmann, Madison and Strandburg2014; Evans et al. Reference Evans, Burke and Jarvik2015). Examples of this approach include the genomic data resources curated by NIH’s ClinGen program and deposited in ClinVar (Rehm et al. Reference Rehm, Berg and Brooks2015). Depositing de-identified data does not require consent of the individual research subjects under current regulations; when identifiable data do need to be shared, the requisite consents can be obtained at the time individuals consent more broadly to participate in the grant-funded research. Another example of incentivized consent alignment is the million-person research cohort under President Obama’s Precision Medicine Initiative, later renamed the NIH All of Us research program, which envisions using a combination of conditional NIH grants to secure cooperation of data-holding research institutions and individual consents/privacy authorization from a cohort of volunteers (Patil, Williams, and Devaney Reference Patil, Williams and Devaney2016).
Publicly funded efforts of this sort have launched the long process of developing national genomic data infrastructure, but they are costly and ultimately not scalable to the task of creating several-hundred-million-person genomic data infrastructures (Evans Reference Evans2016a: 19). To date, the genomic data resources assembled using incentivized consent alignment are small in two respects: first, they include data for only a sliver of the genetic variants that the included individuals have. The NIH ClinVar database just described only holds variants of interest – that is, specific variants that were the focus of a research study or that were related to a disease that led a doctor to order genomic testing. Many and indeed most of the detected variants – such as the variants recorded in individuals’ entire VCF files – are not reported. Existing data resources illuminate small, already explored spaces of the human genome but do little to illuminate the vast dark spaces beyond.
A second problem is that the existing databases are small in the sense of not including data for many people. The FDA’s 2014 discussion paper on NGS proposed the use of databases, including the one “curated by NIH’s ClinGen program and deposited in ClinVar,” to infer the clinical validity of genomic tests (US Dep’t of HHS/FDA 2014c). As of November 2015, despite rapid progress, ClinVar only contained about 130,000 variants that had interpretations of their clinical significance (US Dep’t of HHS/FDA 2015d: 27). For some of those variants, the available “interpretation” consists of a statement that the variant’s clinical significance is unknown (Rehm et al. Reference Rehm, Berg and Brooks2015). To reflect a truly large sample of the population, it ultimately will be necessary to harness data from clinical genetic testing as well as from research studies. The NIH policies that incentivize deposits of data into ClinGen and ClinVar are mandatory only with respect to research data generated using NIH funds. Some commercial clinical laboratories voluntarily contribute data about variants they detect in the course of conducting clinical genomic tests, but others decline to contribute (Evans et al. Reference Evans, Burke and Jarvik2015).
Even if all available genomic data were reported into shared data resources such as ClinVar, the reality is that genomic data – by themselves – are of limited scientific use unless detailed phenotypic data also are available for the same individuals. “Making discoveries” means inferring verifiable associations between gene variants and health conditions, and verifiability means not only that the final scientific conclusion is shared – for example, by sharing a finding that a specific genotype-phenotype association has been detected. It also means that the underlying data that supports the conclusion is available for further inspection and analysis (Cook-Deegan et al. Reference Cook-Deegan, Conley and Evans2013; National Research Council 2003). Existing genomic data commons such as ClinVar typically do not capture detailed phenotypic data, such as a patient’s longitudinal health record or information about lifestyle and environmental factors. Such data would need to come from clinical health care providers, insurers, fitness device manufacturers, and others beyond the reach of NIH’s funding incentives. Moreover, patients would need to consent to the sharing of their entire health histories. Research participants may be willing to allow sharing of specific research data when consenting to a specific research project, but many if not most people would balk at consenting to share their entire cradle-to-grave health histories as a condition of participating in a research project.
4.3 When Consent Alignment Fails
Consent alignment can fail in three ways, if either the individual or the data holder is reluctant to share data or perhaps both are unwilling to share (Evans Reference Evans2016a: 669–74). This section explores existing regulatory and legal pathways that help foster data sharing in these situations. It also identifies key limitations of these approaches.
Quadrant 2 of Figure 4.1 portrays the situation wherein a data holder is willing to share data in its possession, but the data pertains to an individual who does not wish to share (or perhaps cannot be located at the time consent is needed). The Health Insurance Portability and Accountability Act of 1996 (HIPAA, Pub. L. No. 104–191, 1996) Privacy Rule (45 C.F.R. pts. 160, 164) provides pathways that enable sharing data in this situation. The Privacy Rule has various exceptions to its baseline requirement that individuals must authorize disclosures of their data. One of the exceptions is that data holders may share de-identified data without the individual’s authorization (45 C.F.R. § 164.502(d)(2)). Data holders also do not need individual authorization to supply data for use by public health authorities (45 C.F.R. § 164.512(b)(1)(i)). The Privacy Rule also has a waiver provision that lets an Institutional Review Board or Privacy Board (together, IRB) approve the sharing of data for use in research without individual authorization (45 C.F.R. § 164.512(i)).
The Common Rule (45 C.F.R. pt. 46) governs federally funded research and, at some academic institutions, covers all of their research, including privately funded studies. The Common Rule has a waiver mechanism (45 C.F.R. § 46.116(d) of the current regulation that is functionally similar to the one in the HIPAA Privacy Rule. The Common Rule also has definitional nuances that mimic HIPAA’s pathways for supplying data, without consent, in de-identified form or for use in some types of public health activities (Evans Reference Evans2011). The FDA research regulations (21 C.F.R. pts. 50, 56), which apply mainly to commercial research that aims to develop new medical products, resemble the Common Rule in many respects but, importantly, lack a waiver provision. When regulations allow consent waivers or other exceptions to individual consent, these provisions facilitate data sharing in the Quadrant 2 situation. A willing data holder, using these provisions, may contribute data into the genomic data commons on its own initiative, without individual consent.
In the past, these regulatory mechanisms have provided workable access to data for research and public health. Large data holders such as insurers and hospitals often possess data for hundreds, thousands, or even millions of individuals. The regulatory consent exceptions empower data holders to overcome the collective action problems that otherwise would exist at the level of individuals. Each data holder can act as an aggregator of a large data set that includes all of the individuals with which the data holder does business, and the data holder becomes a single point of contact for those seeking data access. The regulatory consent exceptions spell out a narrow set of circumstances (e.g., de-identification of data, public health uses of data, and IRB-approved waivers for research) in which the data holder is granted discretion to share data without individual consents or privacy authorizations. These regulations enable a scheme of data-holder-driven access, in which data holders are the prime movers in assembling large-scale data resources for research and public health.
A classic example data-holder-driven access is FDA’s Mini-Sentinel/Sentinel System, which has been extensively described in the past and is covered in Ryan Abbott’s chapter in this volume (Chapter 6) (Evans Reference Evans2009, Reference Evans2010a, Reference Evans2010b; Mini-Sentinel Coordinating Center 2016; Pharmacoepidemiology and Drug Safety 2012;). As of October 2014, this system had entered voluntary partnerships with 19 data partners – mostly large health insurers and health systems – enabling access to data for 178 million individuals (US Dep’t of HHS/FDA 2015a: 4). Pursuant to the public health exceptions available under the HIPAA Privacy Rule and Common Rule, which applied because Congress authorized FDA to develop this data infrastructure for post-marketing drug safety surveillance, a mere 19 large data holders were able to mobilize data for more than half the individuals in the United States. This demonstrates the power of the traditional, twentieth-century data-holder-driven access scheme, which empowers data holders to act as aggregators to avoid the collective action problems and transaction costs of having to mobilize 180 million people to contribute their data.
Looking to the future, unfortunately, this traditional data-holder-driven scheme is breaking down for reasons explored in detail elsewhere (Evans Reference Evans2016a: 670–71). To summarize the problems, genomic science requires deeply descriptive data sets that link genotypic, phenotypic, lifestyle, and environmental data for each included individual. A single data holder, such as a hospital or insurer, holds data for many individuals, but it only has a small portion of the available data about each of them – for example, a hospital would only have complete records of its own treatment encounters with each individual, and a private health insurer would hold data only for the brief period (on average, about 3 years) that it insures an individual before the individual (or the individual’s employer) shifts to a different plan. Data holders, while well positioned to aggregate data across many individuals, are not well positioned to aggregate data across the multitude of data holders with which each individual has done business.
Another problem is that deeply descriptive data sets – for example, rich data sets that include much of a person’s health history along with genomic and other data – are inherently re-identifiable (see, e.g., Federal Trade Commission 2012). This fact undermines the credibility of traditional regulatory consent exceptions that rely on de-identification as a pretext for unconsented sharing of data. The resulting privacy concerns make it ever more difficult for IRBs to justify the approval of consent waivers, which have been a major pathway of research data access in the past. The traditional scheme of data-holder-driven formation of research data commons, grounded on HIPAA and Common Rule individual consent exceptions that are growing increasingly implausible, functioned fairly well in the past, but it will not suffice as a way to assemble the large-scale, deeply descriptive data resources that the future requires (Evans Reference Evans2016a).
An even more fundamental problem with data-holder-driven access is that commercial data holders, such as clinical laboratories and private health care providers, have various incentives not to share data that they hold (Cook-Deegan et al. Reference Cook-Deegan, Conley and Evans2013). Research laboratories also may hoard data in the hope of preserving their leadership in future discoveries based on the data. The HIPAA and Common Rule consent exceptions allow data holders to share data but do not require them to do so. Commercial clinical laboratories may regard data they generated in the course of their past testing activities as a valuable proprietary asset. In the environment of weakened patent protection after the 2013 Myriad case (133 S.Ct. 2107 (2013)), laboratories may not be able to maintain exclusivity over test administration (the business of offering the test itself). They may, however, be able to maintain a strong competitive advantage if they have previously amassed rich stores of genomic data that enable them to interpret test results more accurately than their competitors are able to do (Cook-Deegan et al. Reference Cook-Deegan, Conley and Evans2013; Evans Reference Evans, McGuire, Kaufman and Curnutte2014c).
Another commercial concern is that data holders face nonzero marginal costs of data preparation and transmittal, as discussed earlier. A 2014 survey by the Health Data Exploration Project found that many mobile and wearable sensor device data holders view the advancement of research as a worthy goal but not their primary business priority (Health Data Exploration Project 2014). Particularly in the genomics and advanced diagnostics industries, innovative companies that hold valuable stores of data sometimes are thinly capitalized start-ups that can ill afford to donate the labor and investments that it would take to link their respective data sets into a shared data infrastructure for research. Data holders also may harbor genuine concerns about patient privacy. Unfortunately, data holders sometimes cite patient privacy concerns as a pretext for hoarding data, even when regulations such as the HIPAA Privacy Rule would allow data to be shared, or, in the alternative, patients might be quite willing to authorize the sharing if asked (but the data holders do not ask them).
Balky data holders create the situation shown Quadrant 3 of Figure 4.1 (Evans Reference Evans2016a: 671–73). Here, the individual may be willing to share her data for use in research, but an uncooperative data holder blocks sharing. The bioethical literature is asymmetrical, evincing deep concern about unconsented data uses (Quadrant 2) while failing to register ethical objections when data holders (or their IRBs) deny access to data for uses of which the individual would have approved (Quadrant 3). In one study, IRBs refused to supply about 5 percent of the requested medical records for a well-documented, congressionally authorized public health purpose, thwarting not only the will of Congress but also of any individuals who may have preferred to participate in the study (Cutrona et al. Reference Cutrona2010).
The HIPAA Privacy Rule provides an access-forcing mechanism that helps address the problem in Quadrant 3. HIPAA grants individuals a broad right of access to their own data held by data holders, such as insurers and most health care providers, that are covered by the Privacy Rule (45 C.F.R. § 164.524). This individual access right is the only provision in the Privacy Rule that requires a HIPAA-covered data holder to disclose data – all other provisions, such as HIPAA’s waiver provision, are permissive (allowing an entity to disclose data when certain conditions are met) but not mandatory. By invoking their Section 164.524 access rights, patients can obtain access to their data, which they then would be free to contribute for research if they so desire.
This important HIPAA access right has recently been extended to cover laboratory-held genomic and other diagnostic information. In 2014, the US Department of Health and Human Services (HHS) amended the Privacy Rule and the CLIA regulations to apply the Section 154.524 access right to HIPAA-covered laboratories, which had not previously been required to comply with it (US Dep’t of HHS/OCR 2014: 7292). An early legal analysis suggested that at genomic testing laboratories, these amendments would allow individuals to obtain not only their final genomic testing reports but also any underlying data that the laboratory maintains in VCF, BAM, or FASTQ files (Evans et al. Reference Evans, Dorschner, Burke and Jarvik2014). During the first year after this new right went into effect, patients reported difficulty obtaining their underlying genomic data from genomic testing laboratories. Early in 2016, however, HHS issued guidance confirming that patients are indeed entitled to receive their underlying genomic data from HIPAA-covered laboratories (US Dep’t of HHS/OCR 2016). HIPAA’s access-forcing mechanism thus can help free genomic data held by uncooperative data holders.
A defect of the Common Rule (including its recent revision) is that it provides no similar access-forcing mechanism to help research subjects obtain access to data about themselves generated during research. Fortunately, many genomic research laboratories are HIPAA-covered entities as a result of being affiliated with larger academic medical centers that have HIPAA-covered status (Evans et al. Reference Evans, Dorschner, Burke and Jarvik2014). If a research laboratory is HIPAA covered, the Section 164.524 access right applies to it, and research subjects should be able to access their data under HIPAA, even though the Common Rule does not respect their right of access to their own data.
By empowering individuals with the power to force access to their own data, the Privacy Rule is positioning them as a force that potentially can drive the formation of genomic data infrastructure in the future. The experience of some disease-specific patient advocacy groups suggests that at least some consumers may be motivated to do so. In general, however, a consumer access right is only a partial solution: once consumers obtain their data, there remains a collective action problem in getting them to work together to develop large-scale genomic data resources. The HIPAA access right thus is only a first step, although it is a crucial one: it can liberate data from uncooperative data holders. The second step – developing institutional arrangements to empower groups of individuals to collaborate to assemble powerful, large-scale health data resources for use in scientific research – presents a different kind of challenge: how to form and operate consumer-driven knowledge commons (see Frischmann, Madison, and Strandburg Reference Frischmann, Madison, Strandburg, Frischmann, Madison and Strandburg2014). This second step is the one where Ostrom’s work on CPRs and other shared resources becomes relevant, as discussed further in the next section.
Before moving on, however, one last failure of consent alignment deserves discussion. In Quadrant 4 of Figure 4.1, neither the data holder nor the individual is willing to share data, leading some commentators to recommend coercive, legislative approaches that would simply place health data into the public domain (see, e.g., Rodwin Reference Rodwin2010). As already noted, the Privacy Rule and Common Rule contain no provisions mandating access to data for research or public health studies. Authority to compel data access must come from other law, such as court orders or state and federal public health laws that require specific types of data to be reported to governmental agencies for particular purposes (Evans Reference Evans2016a: 673–74). One possible alternative would be for Congress to enact legislation requiring a broader program of compulsory data collection to support specific types of genomic research.
With compulsory data-sharing legislation in place, the HIPAA Privacy Rule’s public health exception (45 C.F.R. Section 164.512(b)(1)(i)) would allow covered entities to share data with public health authorities (i.e., to state or federal public health agencies such as the FDA, or to private entities working under contracts with these governmental agencies). HIPAA’s public health exception is broadly worded and allows release of data for a list of activities that includes public health “investigations.” This phrasing suggests public health research as well as routine public health practice activities are permissible grounds for data disclosure (Evans Reference Evans2011). Discovering the clinical significance of the public’s genetic variants clearly would serve an important public health purpose.
All the HIPAA-covered data holder must do is verify three things: (1) that the person requesting the data truly is a public health official (who works for, or is under contract to, a public health authority); (2) that the public health authority really does have legislative authorization to collect the data; and (3) that the data requested is the “minimal necessary” to fulfill the legislatively authorized task – that is, that the official has not requested more data than actually is required for the public health purpose (45 C.F.R. § 164.514). In verifying these three things, a HIPAA-covered entity may rely on the public health authority’s representations. For example, if a public health agency states that it has legislative authorization, the data holder is not required to second-guess that statement. Reliance is permitted so long as it is “reasonable.” Thus, if a person claiming to be a public health official presented a fake-looking badge and credentials, it would be unreasonable to rely on that information and the data holder could face administrative sanctions for releasing the data. If the credentials appear facially reasonable, however, the data holder may respond to a public health official’s request for data without fear of violating the HIPAA Privacy Rule.
A problem with public health legislation of this sort is that compulsory data access raises ancillary legal and ethical issues (Evans Reference Evans2016a: 674). Members of the public have strong feelings about the privacy of their genomic and other health data, and it would be difficult for elected legislators to ignore these strong public sentiments and mandate that all health sector data holders must share people’s data for broad, unspecified genomic research purposes. When legislators pass laws that require health data to be shared, this is usually limited to narrow purposes where the need to gather the data is compelling: for example, tracking epidemics or detecting domestic violence. There arguably is a strong public health rationale for developing large-scale genomic data infrastructure, because genetic diseases – including cancer – potentially affect every member of the public. To date, however, our society has not reached a consensus that the need to unlock the secrets of the human genome is so compelling that it overrides the individual’s interest in data privacy.
Another problem is that forcing private sector data holders to disclose their data holdings may raise takings issues under the Fifth and Fourteenth Amendments to the Constitution, which provide that the government cannot take a person’s property for public use without just compensation (Evans Reference Evans2011). Who owns data held in genomic and health databases is a question of state property law. In most instances, the consumer and the data holder both have various legal interests in the data but, in most states, neither enjoys outright ownership (Evans Reference Evans2011: 73–74). Thus, data holders usually do not have property interests in the data that could support a takings claim. However, data holders may have made significant capital investments and investments of labor to curate and format the data, to convert data into an accessible format, and to develop information systems that render the data useful for clinical and research purposes (Evans Reference Evans2011: 106–07; Evans Reference Evans2014a). Legislation forcing a data holder to share data could diminish the value of those investments, generating takings claims. Even if the government ultimately prevailed on the takings questions, litigation could drag on and delay formation of genomic data commons.
A final point is that compulsory sharing of data would not, in reality, ensure the development of useful genomic data resources. Developing useful data resources requires access not only to data but also to ancillary services that the data holders would need to provide – for example, data preparation services to convert their data into an interoperable format so that it can be analyzed together with data from other sources (Evans Reference Evans2011: 106–7). The government has little power to force unwilling private entities to contribute services (Brenner and Clarke Reference Brenner and Clarke2010), even if data holders could be forced to part with their data. The necessary services can only be procured through consensual methods, such as entering contracts with the data holders or requiring their services as a condition of a grant (Beam and Conlan Reference Beam, Conlan and Salamon2002; Kelman Reference Kelman and Salamon2002). Such methods all would require substantial funding. These considerations all seem to disfavor compulsory data sharing and point to a need for voluntary, incentives-based arrangements (Evans Reference Evans2014a). Ryan Abbott (Chapter 6, this volume) describes the FDA’s use of voluntary approaches in developing its Sentinel System.
The options for developing large-scale genomic data resources can be summarized as follows: consent alignment is a lovely thing when it occurs, but, to date, it has not occurred at the level required to create the massively scaled data infrastructures now needed to move genomic science forward. Incentivized consent alignment, where funding agencies such as the NIH promote data sharing through conditional grants, can jump-start efforts to create genomic data infrastructure, but this approach ultimately is not scalable (Evans Reference Evans2016a: 668–69). Existing regulations such as the HIPAA Privacy Rule and Common Rule enable a scheme of data-holder-driven creation of research data resources that has functioned well in the past but is ill adapted to the challenge of creating large-scale, deeply descriptive data resources to support genomic science (Evans Reference Evans2016a: 669–71). Into this breach, recent amendments to the HIPAA Privacy Rule have expanded individuals’ access to their own data, and this opens a possible new pathway: consumer-driven data commons (Evans Reference Evans2016a: 671–73. However, the success of this pathway will require systematic work to overcome the collective action and governance challenges of getting millions of people to cooperate to create and sustain useful genomic data resources (Evans Reference Evans2016b). A final alternative – legislatively imposed mandatory data sharing – has so many legal drawbacks that it is unlikely to offer meaningful solutions (Evans Reference Evans2016a: 673–74). Surveying this landscape, consumer-driven data commons emerge as the most attractive – albeit far from easy – solution.
4.4 The Emergence of Consumer-Driven Data Commons
A fundamental question is how to conceive the aims of genomic data infrastructure and the role regulatory agencies should play in relation to this infrastructure. One possibility is for genomic data systems to serve as a repository that compiles authoritative records of already discovered associations between genetic variants and health conditions. The database operators would collect discoveries made externally and curate the reported information to reconcile conflicts, if multiple laboratories have assigned divergent clinical meanings to particular genetic variants. The role of a consumer safety regulator, such as FDA, would be limited to certifying the quality of information stored in the database, to assure it provides an authoritative snapshot of what is known about the clinical significance of the genetic variants included at a particular point in time. When approving the safety and effectiveness of new genomic tests, the regulator would allow test manufacturers to make claims about the clinical validity of genomic tests if the database provides support for such claims, and the regulator would block claims that lack database support. This scheme corresponds to the approach FDA proposed in its December 2014 NGS discussion paper (US Dep’t of HHS/FDA 2014c). Such a scheme treats genomic uncertainty as “static” in the sense that the regulator ultimately has no control over it. The regulator can block clinical claims that external sources indicate are too uncertain at a given moment but has no power to hasten the pace at which uncertainty is resolved (Evans et al. Reference Evans, Burke and Jarvik2015).
An alternative view is that genomic data commons should be interoperable information systems capable of supporting fresh discoveries of as-yet-unknown associations between genetic variants and human health. By this view, genomic databases are not mere repositories of externally discovered associations but instead should serve as critical resources to fuel an active, ongoing, continuous discovery process involving a wide range of academic, commercial, and public health researchers, all of whom contribute to the grand challenge of deciphering the clinical meaning of the human genome. This scheme would treat uncertainty about the clinical significance of gene variants as “dynamic” in the sense of being a parameter that regulatory policies can influence. One of the regulator’s major responsibilities would be to implement policies that hasten the pace at which uncertainty shrinks through the discovery of new, statistically significant relationships between genotypes and phenotypes (Evans et al. Reference Evans, Burke and Jarvik2015). In this scheme, the regulator is not merely an information taker but instead is an information steward. Jorge Contreras (Chapter 2, this volume) explores the regulator’s role in further detail.
Existing genomic data infrastructures, such as ClinGen/ClinVar and the cohort planned as part of the Precision Medicine Initiative/All of Us research program, are moves in the right direction, but they have not yet achieved the scale and the level of detail that ultimately will be required. Financial sustainability is a major concern with systems such as these, financed with limited-term grants and other sources of federal funding (Evans et al. Reference Evans, Burke and Jarvik2015). Sustainability will require a workable revenue model to cover system costs, including such items as the costs data holders incur as they prepare and transmit data to the system, costs of developing and maintaining the system’s information infrastructure, and costs of ensuring data security protections and rigorous ethical/legal compliance.
The United States has a long history, dating back to the late 1800s, of mobilizing private capital to help develop major national infrastructure such as railroads, high-voltage power transmission grids, natural gas pipeline networks, and telecommunications infrastructure (Frischmann Reference Frischmann2012). Similar voluntary, incentive-based approaches may offer promise in genomics (Evans Reference Evans2014a, Reference Evans, McGuire, Kaufman and Curnutte2014c). The genomic data resource system should be viewed as one of a long line of national infrastructure challenges that many nations have faced in the past and continue to face in the twenty-first century (Frischmann Reference Frischmann2012).
Before policymakers embrace any approach, however, a crucial decision node often goes unacknowledged. Any voluntary, incentive-based approach must first establish who are the volunteers it seeks to incentivize. Control of genomic and other relevant health data, as portrayed in Figure 4.1, is fragmented at two levels: at the level of data holders and at the level of individual data subjects. One alternative – for convenience, a data-holder-driven strategy – would direct incentives toward institutional data holders who can, if suitably motivated, invoke various consent exceptions in privacy and human-subject protection regulations to facilitate flows of individual data into genomic data commons. The other alternative – a consumer-driven strategy – would direct incentives toward individual data subjects, who can invoke the access-forcing mechanism in existing privacy law to free their data from recalcitrant data holders and contribute the data to form useful genomic data infrastructures. For reasons outlined in the previous section, this latter approach appears to be the last strategy standing that has a potential to work, but it will require considerable effort going forward, because institutional arrangements are not yet in place to orchestrate the required levels of collective action.
An important point to emphasize is that data-holder-driven and consumer-driven strategies are equally viable from a legal standpoint. Policymaking efforts of the past – and most recent proposals – have focused on data-holder-driven strategies that treat data holders as the prime movers of data infrastructure development, while dismissing consumer-driven strategies. The idea of engaging individuals in the creation of genomic data commons was raised at FDA’s November 2015 Public Workshop exploring use of databases to establish clinical validity of genomic tests:
Across the country and the various states patients are getting more and more access to not only their clinical tests, their medical records but also their lab test records. Quite often in many cases it is the patient that is most incentivized to find out what they can about their condition. What does the panel think about taking crowd sourcing to patient level?
This suggestion drew skepticism, primarily reflecting concerns that patient-handled data would be prone to errors and inconsistencies (US Dep’t of HHS/FDA 2015d: 40–43). One response to such concerns is that data from all health care sources is prone to errors and inconsistencies; scrubbing and reformatting data into a common data format is always one of the most challenging and time-consuming components of any effort to create useful health data systems, regardless of the data source (Evans Reference Evans2011; P-CAST 2010: 39). Information in electronic health records held by health care providers and payers is often directed toward billing, which introduces its own biases and inaccuracies (US Dep’t of HHS/FDA 2015d: 71), such as a tendency to overstate the gravity of a patient’s symptoms to qualify for a higher insurance reimbursement, or the tendency to record that a diagnostic test was performed without bothering to note what the test discovered (Evans Reference Evans2010b: 483). Another response is that HIPAA’s Section 164.524 access right, which allows individuals to free their data held by HIPAA-covered laboratories and health care providers, also allows them to direct that the data be transmitted directly on their behalf to a third party, which could be a qualified data infrastructure operator. Thus, data do not need to pass through individuals’ hands while being conveyed, at their instruction, to the genomic data resource system. This could help allay concerns that individuals may alter or corrupt information that passes through their hands.
Despite the skepticism expressed at FDA’s Public Workshop, the idea of involving consumers in genomic data infrastructure development gained some support, with experienced geneticists noting that patients often have phenotypic information beyond what is in their medical and insurance records, and that this information is “incredibly accurate in many cases” (US Dep’t of HHS/FDA 2015d: 70). The discussion ended with suggestions to “embrace” patient-driven strategies more (US Dep’t of HHS/FDA 2015d: 71).
How to mobilize consumer-driven strategies is the unanswered question, and this is not a question that this chapter can fully answer, except by noting that early work already is under way (Evans Reference Evans2016a, Reference Evans2016b) to position consumer-driven genomic data commons in the analytical framework associated with Elinor Ostrom and her modern interpreters (Frischmann, Madison, and Strandburg Reference Frischmann, Madison, Strandburg, Frischmann, Madison and Strandburg2014; Hess and Ostrom Reference Hess and Ostrom2006; Ostrom Reference Ostrom1990). This framework offers intriguing insights. To cite one example, Ostrom’s exploration of self-organizing and self-governing CPRs suggests principles, such as the use of nested enterprises involving small groups that elicit trust (Ostrom Reference Ostrom1990: 189). This approach for overcoming collective action problems evokes a pattern of activity already observed among disease-specific patient advocacy groups that have created disease-specific data resources, such as a cystic fibrosis genetic database that already is supplying reliable data to support FDA review of genomic tests (US Dep’t of HHS/FDA 2015d: 16). The Precision Medicine Initiative apparently has embraced a centralized data architecture for its one-million-person cohort study, and a centralized database is a feasible alternative at such a small scale – and make no mistake: one million is small scale in the context of genomic research. Genomic data resource systems ultimately must attain a much larger scale, and a distributed network-of-networks architecture, such as the nested enterprise model Ostrom suggests, may have merit, particularly in overcoming the reluctance of individuals to cede their data to a centralized network operated by strangers.
Early work on consumer-driven data commons conceives these entities as institutional arrangements to empower groups of consenting individuals to collaborate to assemble powerful, large-scale health data resources for use in scientific research, on terms the group members themselves would set (Evans Reference Evans2016a, Reference Evans2016b). The consumer-driven data commons movement is, above all, a critique of the atomistic vision of individual autonomy that pervaded twentieth-century bioethics: a presumption that patients, research subjects, and consumers of direct-to-consumer health-related services are fundamentally alone, individualistic, disorganized, weak, and vulnerable (Tauber Reference Tauber2005: 13, 117) and in need of paternalistic protectors – concerned bioethicists and dedicated IRBs – to look after their interests. This presumption of human disempowerment pervades federal regulations such as the HIPAA Privacy Rule and Common Rule, which reject the approach of organizing individual consumers to protect themselves, for example, by unionizing them to protect their own interests through collective bargaining with researchers who want to use their data (Evans Reference Evans2016b). The federal regulatory framework that set out to protect individuals ultimately disempowered the very patients and research subjects it sought to protect, empowering them to make decisions as individuals, but only as individuals, and lacking a roadmap for collective action (Evans Reference Evans2016b: 248).
Edmund S. Morgan has chronicled developments, in the 1600s and 1700s, that erupted in the history-altering concept that individual British subjects are not fundamentally alone and limited to one-on-one autonomous interactions with the king in whose presence they are weak and disempowered. Instead, individuals can interact with one another to create a larger fictional construct, The People, which exudes “a certain majesty [that] could be placed in the scales against the divinity of the king” (Morgan Reference Morgan1988: 24). Consumer-driven data commons are institutional arrangements to bring about a similar moment of truth for traditional bioethics: those whose data populate large-scale health data networks are awakening from the condition Thomas Hobbes referred to as “the confusion of a disunited multitude,” unifying into a People that rejects the privacy and access standards of the past and demands new standards of the people, by the people, and for the people (Evans Reference Evans2016b: 252; Hobbes Reference Hobbes1952).
Surveys show that a majority – up to 80 percent –of consumers have a positive attitude about health-related research and support the concept that their health data should be used to advance research (Health Data Exploration Project 2014; Kish and Topol Reference Kish and Topol2015; Topol Reference Topol2015). Yet very few people actually volunteer their data for research (Evans Reference Evans2016a: 683; Evans Reference Evans2016b: 247). The inescapable conclusion is that people are not comfortable with the ethical and privacy frameworks that bioethical experts and regulators have devised, top-down, to protect them. Consumer-driven data commons would engage consenting individuals in the task of designing better ethical and privacy frameworks to govern access to their data.
Twentieth-century genomic science requires large data sets aggregated across many individuals and many data holders. Traditional data-holder-driven data access mechanisms are good at aggregating data across people, but not across data holders. Individual health care consumers now have the power to aggregate data about themselves across many data holders by using their HIPAA access rights. But can they work together to aggregate their data across large, diverse groups of individuals? That is the question.
The vision of consumer-driven data commons is that individuals would obtain their own health data by exercising their HIPAA Section 164.524 access rights and voluntarily deposit their data into one or more consumer-driven data commons (Evans Reference Evans2016b: 262–64). No individual would be required to join such a group. Each consumer-driven commons would announce transparent rules addressing how to become a member of the group, the duties of membership, and the terms of exit from the group. Members of each commons would decide, through self-governance processes, which research uses of their collective data resources are permissible and which are not. These collective decisions would be binding on the members for so long as they choose to remain in the commons. Thus, commons groups would be able to offer high-valued, aggregated data sets that incorporate deeply descriptive longitudinal data for their entire membership (Evans Reference Evans2016b: 262-64).
Individual members, when joining a consumer-driven commons, would cede their right of traditional, individualized informed consent. Instead, they would agree to be governed by the collective decision-making processes of the group, in which they would have a meaningful voice. Thus, each individual’s right of informed consent would be exercised at the point of making a decision to join, stay in, or leave a particular consumer-driven data commons group. While in it, individuals in effect would appoint the group’s decision-making body to act as their personal representative (as is allowed by the HIPAA Privacy Rule and Common Rule) to consent to specific uses of their data on their behalf. These commons-forming groups could be membership organizations organized by data-contributing individuals themselves, or they could be organized by disease advocacy groups or by commercial data management companies that agree to manage members’ collective data resources according to rules the members themselves would set (Evans Reference Evans2016b: 262–64).
What are the advantages of consumer-driven data commons? The greatest advantage is the power of data aggregation and collective decision making. An individual, acting alone, can almost never (unless she has an incredibly bizarre and fascinating medical condition) offer a data resource that is sufficiently attractive to place the individual in a position to set terms and conditions about how the data can be used, what privacy protections should apply, and what data security protections are expected. The right of individual informed consent granted by regulations such as the Privacy Rule and Common Rule is basically a useless, take-it-or-leave-it right: individuals can consent to privacy and ethical practices defined by regulations and by the data user, or they can take their data and go home, but they cannot negotiate the terms on which their data can be used (Evans Reference Evans2016b: 248). The aim of consumer-driven data commons is to place groups of like-minded individuals in a position to insist that their collective, well-considered privacy and ethical preferences be given weight. A large group of individuals who deposit their data into a consumer-driven data commons can amass a sufficiently attractive data resource to make demands.
As commons-forming groups enunciate their respective visions of ethical data access, a “marketplace” of ethical and privacy policies would emerge (Evans Reference Evans2016b: 264). Individuals could compare these policies when choosing which consumer-driven data commons to join. Successful consumer-driven commons would be the ones that offer policies that address the concerns that data-contributing individuals feel, while still making data available for useful lines of research that benefit their members and the public at large. As these successful consumer-driven data commons expand, their policies would inform the policies of other commons-forming groups, leading to a possible convergence of consumer-driven bioethical standards to replace the top-down, expert-driven bioethical standards in place today (Evans Reference Evans2016a: 683).
Another crucial advantage of consumer-driven data commons is that they may be better positioned to achieve financial sustainability than data-holder-driven commons are under current US law. Federal regulations such as the HIPAA Privacy Rule do not restrict individuals’ ability to sell their own data (Evans Reference Evans2016a: 681). Consumer-driven data commons – assuming their members are comfortable with the ethics of data commodification – would be able to charge data access fees to cover the costs of engaging suitably skilled consultants to convert their members’ data into interoperable formats, to develop system infrastructure, and to operate their collective data resources on an ongoing basis.
In contrast, institutional data holders are subject to restrictions under the 2009 Health Information Technology for Economic and Clinical Health (HITECH) Act (Pub. L. 111–5, Div. A, Title XIII, Div. B, Title IV, 123 Stat. 226, 467 (Feb. 17, 2009)). HITECH inserted data sales restrictions into the HIPAA Privacy Rule that do not allow HIPAA-covered entities to sell data (42 U.S.C. § 17935(d)(1); 45 C.F.R.§164.502(a)(5)(ii)). They can, however, charge a reasonable, cost-based fee for data preparation and transmittal services when they share data for research under the Privacy Rule’s waiver provision (45 C.F.R. §164.502(a)(5)(ii)(B)(2)(ii)). This cost-based fee, had it been implemented properly, could have supported robust data sharing, just as cost-of-service utility rates have successfully supported the development of major national infrastructures in many industrial sectors (Evans Reference Evans2014a, Reference Evans2014b). Unfortunately, the 2013 Privacy Rule revisions implementing the cost-based fee set it so low that it denies data holders a reasonable rate of return on capital they invest in data development and system infrastructure (US Dep’t of HHS/OCR 2013). This limits data holders’ potential to lead the charge in developing the genomic data resources that twenty-first-century science needs. As explained elsewhere, the current regulations have the unintended consequence of virtually forcing data holders to embrace distributed network architectures if they wish to earn a reasonable rate of return on their invested capital (Evans Reference Evans2014a: 508, col. 2). Distributed data architectures have many merits, but decisions about system architecture should be guided by system performance objectives rather than turning on accidents in regulated pricing formulas. Consumer-driven data commons, unburdened by the current restrictions on cost recovery, appear better positioned to pioneer financially sustainable data sharing solutions.
Conclusion
Consumer-driven genomic data commons, at this point, are in the earliest phases of conceptualization and should be viewed “not as a panacea, but as a grand experiment in democratic data self-governance, perhaps worth trying at a time when existing mechanisms of data access seem destined not to meet the challenges that lie ahead” (Evans Reference Evans2016a: 684). The goal of this chapter was to describe the legal obstacles that stand in the way of developing large-scale genomic data resources and to invite the community of commons scholars to get involved with overcoming them.
The ultimate value of Ostrom’s work, in the context of genomic testing, may lie in its potential to reframe the human genome as a shared resource that can and should be managed through collective efforts of the people whose data are involved. Genetic information is often characterized as deeply personal and private, but this is a mistake: to test one’s genome is to enter a public space where one discovers what one has in common with other people. Gene variants that are unique can never have an established clinical validity because clinical validity can be inferred only by comparing an individual’s variants to those of other people. We shall “crack” the human genome and discover its meaning together, or not do so at all.
The appropriate norm, as we undertake this challenge, is the norm of “common purpose” recently enunciated by Ruth Faden and her coauthors (Faden et al. 2013). They acknowledged that the moral framework for twenty-first-century science may differ in significant respects from traditional conceptions of clinical and research ethics” and may include an obligation for individuals to participate in knowledge-generating activities (Faden et al. 2013: S16, S18). They see this as a bounded obligation that would vary with the degree of risk and burden involved, so that individuals would not be obligated to participate in clinical trials that pose physical risks but may have an obligation to contribute their data to studies that offer the prospect of useful scientific advances (Faden et al. 2013: S23). They suggest that this obligation is grounded in a “norm of common purpose … a principle presiding over matters that affect the interests of everyone” (Faden et al. 2013: S16): “Securing these common interests is a shared social purpose that we cannot as individuals achieve” (Faden et al. 2013: S16).
The unanswered questions with this common purpose ethical framework are how to operationalize it, monitor it, and overcome the collective action and governance problems it presents. Ostrom’s case studies of community efforts to create self-organized and self-governing commons in other contexts may provide the missing piece that bioethicists have as yet been unable to supply: a set of principles drawn from how real people in real circumstances have been able to create sustainable commons.


 Open access
Open access